 2018, Vol. 29
2018, Vol. 29 



b School of Chemistry and Chemical Engineering, Liaocheng University, Liaocheng 252000, China;
c School of Chemistry and Chemical Engineering, Hebei Normal University For Nationalities, Higher Education Park, Chengde 0670000, China
Catalytic enantioselective formation of C-Si bonds has attracted much attention over the past decade due to the versatility of the resultant optically active silylated compounds [1]. Therefore, several methods have been developed by using chiral transition metal complexes as catalysts and (dimethylphenylsilyl)boronic acid pinacol ester (PhMe2Si-Bpin) was usually employed as silylating reagent for these silyl transfer reactions [2]. Moreover, optically active α-hydroxysilanes, a class of important chiral organosilicone compounds, have been widely used in the formation and rearrangements of stereocontrolled C-C bond, which results in a variety of chiral organic compounds [3]. Thus, development of practical and efficient methods for catalytic enantioselective preparation of chiral α-hydroxysilanes is an important and challenging goal of research in chemical synthesis. As we all know, the asymmetric reduction of acylsilanes [4] and the hydrogenation of enolsilanes [5] are the majority of methods for the preparation of chiral α-hydroxysilanes, which suffers from the requirement of multistep reactions [6] and a lack of functionality tolerance owing to using strong bases. In their pioneering work, Kleeberg and Oestreich reported the racemic 1, 2-addition of a silicon nucleophile to aldehydes catalyzed by a carbene–Cu complex and proposed a reasonable catalytic cycle [7]. The first example of Cu-catalyzed enantioselective 1, 2-addition of a silicon nucleophile to aldehydes was reported by Riant et al. in [8]. Then our group reported a transition metal-free method for enantioselective silyl addition to aromatic aldehydes in a mixture of tetrahydrofuran and methanol [9]. Despite their potential utility, existing methods are not environmentally friendly and suffer from one or more disadvantages, for example, hazardous organic solvents, complex workup and purification, use of toxic metal catalysts, poor yields, and long reaction time. Therefore, the development of simple, safe, economical and environmentally friendly methods for the preparation of chiral α-hydroxysilanes is still a challenge.
Exploring organic reactions in water is very important in modern organic chemistry because water is non-flammable, nontoxic and non-carcinogenic, and in addition, water is probably the least expensive and most easily accessible solvents. Moreover, running a reaction in water instead of an organic solvent leads to novel modes of transformations and produces unique reactivity and selectivity [10]. Indeed, the catalytic reactions sometimes exhibit better performance in water over analogous homogeneous systems [11].
The coenzyme thiamine (vitamin B1, a natural thiazolium salt) is involved in many enzymatic processes, in which the catalytically active species has been proposed to be a carbene. Great efforts have been made to perform enzyme mimetic asymmetric carbene catalysis [12]. Among the N-heterocyclic carbenes, [2.2]paracyclophane backbone is one of the most important scaffolds due to its utility as a planar chiral source in asymmetric catalysis [13]. Herein, we disclose our findings that N-heterocyclic carbenes derived from [2.2]paracyclophane are effective catalysts for the enantioselective 1, 2-silylation of aromatic aldehydes in water without the use of any harmful organic solvents and transitionmetal salts.
It is well known that solvents play a key role in many organic reactions as they modify both reaction equilibrium and kinetics. To our surprise, in carbene-catalyzed silyation reaction [9, 14, 16], it has been shown that water or methanol can bear other functions such as acting as promoter and even co-catalyst beyond their role as reaction media. These results indicate that the proton sources (water or methanol) are required for efficient and enantioselective silyl addition reaction. Therefore, we designed two new hydroxylfunctionalized N-heterocyclic carbene precursors derived from [2.2]paracyclophane (Scheme 1). We anticipated that a hydroxyl group would be introduced into the [2.2]paracyclophane skeleton using a Suzuki-Miyaura cross coupling approach. As expected, 2a and 2b were obtained in high yields by Pd-catalyzed crosscoupling reaction of enantiomerically pure 1 [14] with aromatic boronic acids. On the basis of the previous reports [15], (S, Sp)-4 and (S, Sp)-5 were easily prepared in two steps from 2a and 2b in 66% and 85% yields, respectively (Scheme 1).

|
Download:
|
| Scheme 1. Synthesis of N-heterocyclic carbene precursors | |
According to the procedure of Hoveyda for the carbenecatalyzed asymmetric conjugate addition of a silyl group to α, β-unsaturated ketones [16a][16a], we first probed whether the new hydroxyl-functionalized carbene precursor (S, Sp)-4 could catalyze the silylation of aromatic aldehydes efficiently and enantioselectively (Table 1). Fortunately, with 5.0 mol% (S, Sp)-4, 30 mol% of DBU, 1.0 equiv. of benzaldehyde, and 1.5 equiv. of Me2PhSiBpin in a solution that largely consists of water (THF:H2O = 1:3), the desired product 7a was obtained in moderate yield and enantioselectivity without the formation of benzoin 7b (Table 1, entry 1). However, the silyl addition did not take place to any appreciable extent in an equal mixture of THF and water (Table 1, entry 2). Moreover, the above reactions did not proceed when performed in typical organic solvents (Table 1, entries 3–6). These results suggest that the amount of water has a significant influence on the reaction rate, and the use of pure water as solvent might be a viable solvent so as to compensate for the lack of reactivity. Just as expected, the silylation reaction proceeded more smoothly in pure water than in a mixture of THF and water, so pure water was chosen as the optimal solvent (Table 1, entry 7). To improve the yield and enantioselectivity, we then explored the reaction parameters with a particular emphasis on base effects. Unfortunately, the yield decreased dramatically by using N, N-diisopropylethylamine instead of DBU (Table 1, entry 8). Next, some water-soluble bases were used for the reaction, such as NaOAC, KF and NaOH. When NaOAc was subjected to the silyl transfer reaction, the reaction did not occur (Table 1, entry 9). It was observed that KF or NaOH exhibited much higher reactivity but a slightly lower level of enantioselectivity was obtained (Table 1, entries 10–11). Subsequently, the impact of the amount of base was also evaluated. Reduction of the amount of NaOH from 30 mol% to 5 mol% caused a sharp decrease in the reactivity (Table 1, entry 12). In light of the negative effect of NaOH, we turned our attention back to DBU. To our delight, the highest yield was achieved by increasing the amount of DBU from 30 mol% to 100 mol% without affecting the enantioselectivity, indicating that 100 mol% DBU was the most suitable choice (Table 1, entry 13). Interestingly, as shown in Table 1, changing the reaction conditions could not alter the enantioselectivity by an appreciable amount.
|
|
Table 1 Screening of the reaction conditions |

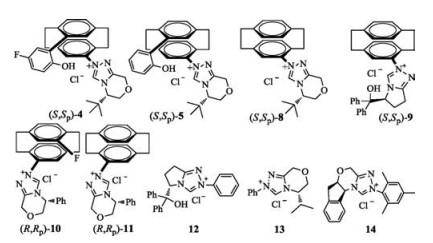
As the catalytic activity reached an acceptable level, we focused our attention on identifying an effective chiral carbene catalyst under the optimal reaction conditions (Fig. 1). In the beginning, the asymmetric silyl addition of benzaldehyde was performed at 25 ℃ in water by using an analogous [2.2]paracyclophane-based triazolium bearing a pseudo-ortho substituted phenol moiety ((S, Sp))-5 as carbene precursor. In comparison to the above used fluorine-substituted (S, Sp)-4, chiral triazolium salt (S, Sp)-5 showed a lower catalytic activity but the enantiomeric ratio value remained the same (Table 2, entries 1 and 2). It should be noted that OH function in para-fluorophenol-substituted (S, Sp)-4 is effectively more acidic than phenol-substituted (S, Sp)-5 containing OH function, which could result in the more efficient carbeneborosilane complex generation and more rapid carbene catalyst regeneration [16b]. This could explain the good yield observed with fluorine-substituted (S, Sp)-4.

|
Download:
|
| Fig. 1. Structures of chiral carbene precursors used in this work | |
|
|
Table 2 Examination of chiral NHC catalysts |

To clarify the effects of hydroxyl-functionalized carbene precursor (S, Sp)-4 on the yield and enantioselectivity, we carried out the same reaction with unsubstituted [2.2]paracyclophanebased triazolium salt (S, Sp)-8. It was found that unsubstituted [2.2] paracyclophane-based triazolium salt (S, Sp)-8 exhibited a lower catalytic activity in terms of either yield or steroselectivity (Table 2, entry 3). In contrast, hydroxyl-functionalized triazolium salt (S, Sp)- 9 derived from L-pyroglutamic acid, which induced exceptional enantioselectivities in Cu-catalyzed β-boration of α, β-unsaturated carboxyl compounds [15c], promoted not only C-Si bond formation but also benzoin condensation with moderate efficiency, led to a 54:33 mixture of 7a and 7b in 87% yield. Furthermore, the enantiomeric ratio value for 7a was only 56:44 (Table 2, entry 4). The use of (R, Rp)-10 and (R, Rp)-11 for the silyl transfer reaction gave 7a as a major product in moderate yield (45% and 43%, respectively) but with a much lower enantioselectivity in both cases (Table 2, entries 5 and 6). These results revealed that the structure of the substituent at central chiral carbon atom in the triazolium motif had a stunning effect on yield and enantioselectivity, indicating that the tert-butyl group was the most suitable substituent for performing the desired 1, 2-silyl addition. Then, we extensively screened several commercially available chiral triazolium salts which were widely applied in organocatalytic enantioselective transformations. However, no products can be detected by using chiral carbene precursors 12 and 13 as catalysts (Table 2, entries 7 and 8). When chiral aminoindanol derived triazolium salt 14 was used for the reaction under the same reaction conditions, not surprisingly, benzoin 7b instead of the silyl adduct 7a was obtained in 94% yield (Table 2, entry 9). Based on the research findings, we can conclude that planar chiral [2.2]paracyclophane moiety in the triazolium frame plays a key role in the carbenecatalyzed asymmetric 1, 2-silylation. We surmised that more electron-donating [2.2]paracyclophanyl substituent increased the Lewis basicity of the carbene catalyst and facilitated formation of a more reactive carbene–borosilane complex which resulted in a more efficient silyl addition. Furthermore, the screening of carbene precursors revealed that (S, Sp)-4 was still the optimal catalyst in terms of either yield or stereoselectivity.
With the optimal conditions in hand, we examined the scope of the silylation with various aromatic aldehydes containing diverse steric and electronic properties. Surprisingly, the reactions with a series of derivatives of benzaldehyde bearing different substituents such as CN, Br, Cl, CH3 and CF3 proceeded much more slowly than that found in the benzaldehyde case. The lower reactivity can be explained by the solubility behavior of starting materials, that is, substituted benzaldehyde is more lipophilic than benzaldehyde which caused the different degree of phase separation for each substrate and catalyst, thus lowering the reactivity. The stirring speed was changed from 300 rpm to 1000 rpm, however, did not lead to significant improvement in reaction outcomes. It is wellknown that the miscibility of the phases can be enhanced by increasing the reaction temperature. Accordingly, by heating the reaction mixture, the appropriate amount of water can be transported into the organic layer that is sufficient to generate enough BPin hydrolysis for efficient silyl transfer reaction. In line with our expectations, by increasing reaction temperature from 25 ℃ to 60 ℃, the reactions with a series of derivatives of benzaldehyde were found to proceed smoothly in water, even when the substrates are solid. Thus, various aromatic aldehydes including those having electron donating and electron withdrawing substituent at different positions on the phenyl ring, as well as naphthalene derivative were compatible with the new reaction conditions, and provided the corresponding products in good to high yields albeit with moderate enantioselectivities (Table 3). Furthermore, increasing reaction temperature resulted in a somewhat lower enantioselectivity (Table 3, entry 1). Next, we continued to evaluate various substituents at different positions in the phenyl ring in order to investigate the substituent effects. It was found that the substrate bearing a substituent at the para-position on the benzene ring provided the corresponding product with higher enantiomeric ratio than that bearing a substituent at the ortho- or meta-position (Table 3, entries 2 and 12). Moreover, aromatic aldehyde with an electron donating group gave the corresponding silyl adduct in lower yield than that with an electron withdrawing group except for the para-trifluoromethyl benzaldehyde, in which the yield was only 76% (Table 3, entries 2– 12). However, an extended aromatic ring such as naphthalene was not a good substrate and afforded the desired product in lower yield, even though the reaction time was prolonged to 36 h (Table 3, entry 13). Unfortunately, aliphatic aldehydes such as butyl aldehyde and tert-butyl aldehyde were unreactive under the same reaction conditions (Table 3, entries 14 and 15). It was found that the absolute configurations of the major enantiomers of 7a–7n were determined to be (S) by comparison of the specific rotation with the literature data [9].
|
|
Table 3 Investigating the substrate scope of the reaction |
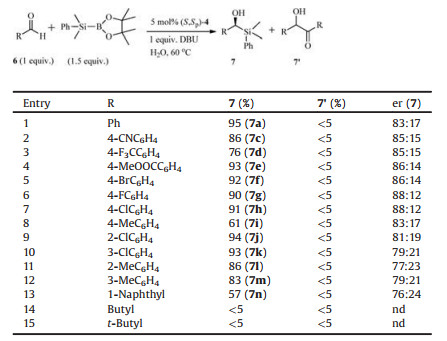
{kind=link}
{kind=link}
Based on the present results and the previous literature [16b], a plausible reaction mechanism was proposed in Scheme 2. The reaction of in-situ-generated carbene (A) with a more sterically accessible borosilane produced by rapid hydrolysis of the sterically demanding BPin unit with excess DBU in water, results in the formation of a more reactive carbene–borosilane complex (B) which is critical for activation of the B–Si bond. The addition of polarized carbene–borosilane complex (B) to the Si-face of the substrate carbonyl group leads to complex (C), which suffers from a lower degree of steric repulsion between the [2.2]paracyclophane moiety, the tert-butyl group of the chiral carbene and the Ar substituent of the aromatic aldehyde, accounting for the stereochemical outcome of the reaction.

|
Download:
|
| Scheme 2. A plausible reaction mechanism | |
{kind=link}
In summary, a new hydroxyl-functionalized triazolium salt based on the [2.2]paracyclophane backbone were synthesized and successfully applied to enantioselective silylation of aromatic aldehydes, affording the corresponding chiral α-hydroxysilanes in one-step. To our knowledge, this is the first example of metal-free enantioselective silylation of aromatic aldehydes in water, which might provide a new direction to the design of the chiral carbene backbone for sustainable reaction processes. It is worth noting that water was not only employed as a reaction medium, but also played a key role in accelerating the silyl transfer reaction. Furthermore, the reaction was performed in pure water, which was easier to operate than the transition metal-catalyzed transformations and proceed without the need for any harmful organic solvents and transition-metal salts.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (Nos. 21372144, 81473085) and Department of Science and Technology of Shandong Province is gratefully acknowledged.
| [1] |
T.H. Chan, D. Wang, Chem. Rev. 92 (1992) 995-1006. DOI:10.1021/cr00013a012 |
| [2] |
(a) T. Mita, M. Sugawara, K. Saito, Y. Sato, Org. Lett. 16(2014) 3028-3031; (b) K. Lee, A. H. Hoveyda, J. Am. Chem. Soc. 132(2010) 2898-2900; (c) M. Takeda, R. Shintani, T. Hayashi, J. Org. Chem. 78(2013) 5007-5017; (d) V. Pace, J. P. Rae, H. Y. Harb, D. J. Procter, Chem. Commun. 49(2013) 5150-5152; (e) C. Zhao, C. Jiang, J. Wang, Asian J. Org. Chem. 3(2014) 851-855; (f) M. Oestreich, E. Hartmann, M. Mewald, Chem. Rev. 113(2013) 402-441. |
| [3] |
(a) I. Fleming, A. Barbero, D. Walter, Chem. Rev. 97(1997) 2063-2192; (b) J. R. Huckins, S. D. Rychnovsky, J. Org. Chem. 68(2003) 10135-10145; (c) P. A. Jacobi, C. Tassa, Org. Lett. 5(2003) 4879-4882; (d) S. Perrone, P. Knochel, Org. Lett. 9(2007) 1041-1044. |
| [4] |
(a) K. Takeda, Y. Ohnishi, T. Koisumi, Org. Lett. 1(1999) 237-239; (b) N. Arai, K. Suzuki, S. Sugisaki, H. Sorimachi, T. Ohkuma, Angew. Chem. Int. Ed. 47(2008) 1770-1773; (c) J. I. Matsuo, Y. Hattori, H. Ishibashi, Org. Lett. 12(2010) 2294-2297; (d) P. A. Jacobi, C. Tassa, Org. Lett. 5(2003) 4879-4882. |
| [5] |
(a) L. Panella, B. L. Feringa, J. G. de Vries, A. J. Minnaard, Org. Lett. 7(2005) 4177-4180; (b) S. Bain, S. Ijadi-Maghsoodi, T. J. Barton, J. Am. Chem. Soc. 110(1988) 2611-2614. |
| [6] |
(a) P. C. Bulman, S. S. Klair, S. Rosenthal, Chem. Soc. Rev. 19(1990) 147-195; (b) R. B. Lettan Ⅱ, B. C. Milgram, K. A. Scheidt, Org. Synth. 84(2007) 22-31. |
| [7] |
C. Kleeberg, E. Feldmann, E. Hartmann, D.J. Vyas, M. Oestreich, Chem. Eur. J. 17 (2011) 13538-13543. DOI:10.1002/chem.v17.48 |
| [8] |
V. Cirriez, C. Rasson, T. Hermant, et al., Angew. Chem. Int. Ed. 52 (2013) 1785-1788. DOI:10.1002/anie.v52.6 |
| [9] |
P. An, Y. Huo, Z. Chen, C. Song, Y. Ma, Org. Biomol. Chem. 15 (2017) 3202-3206. DOI:10.1039/C7OB00243B |
| [10] |
(a) T. Kitanosono, L. Zhu, C. Liu, P. Xu, S. Kobayashi, J. Am. Chem. Soc. 137(2015) 15422-15425; (b) S. Kobayashi, P. Xu, T. Endo, M. Ueno, T. Kitanosono, Angew. Chem. Int. Ed. 51(2012) 12763-12766. |
| [11] |
(a) M. O. Simon, C. J. Li, Chem. Soc. Rev. 41(2012) 1415-1427; (b) G. K. Min, D. Herna'ndez, T. Skrydstrup, Acc. Chem. Res. 46(2013) 457-470. |
| [12] |
(a) D. M. Flanigan, F. Romanov-Michailidis, N. A. White, T. Rovis, Chem. Rev. 115(2015) 9307-9387; (b) X. Wu, L. Hao, Y. Zhang, Angew. Chem. Int. Ed. 56(2017) 4201-4203; (c) X. Chen, K. Chen, D. Sun, S. Ye, Chem. Sci. 8(2017) 1936-1941; (d) K. Zhao, D. Enders, Angew. Chem. Int. Ed. 56(2017) 3754-3756; (e) G. Li, Q. Gu, S. You, Chem. Sci. 6(2015) 4273-4278; (f) J. Chen, P. Yuan, L. Wang, Y. Huang, J. Am. Chem. Soc. 139(2017) 7045-7051; (g) J. Xu, S. Zheng, J. Zhang, X. Liu, B. Tan, Angew. Chem. Int. Ed. 55(2016) 11834-11839; (h) G. Zhang, S. Yang, X. Zhang, et al., J. Am. Chem. Soc. 138(2016) 7932-7938; (i) J. Wu, C. Zhao, J. Wang, J. Am. Chem. Soc. 138(2016) 4706-4709. |
| [13] |
(a) X. L. Hou, X. W. Wu, L. X. Dai, B. X. Cao, J. Sun, Chem. Commun. (2000) 1195-1196; (b) X. W. Wu, X. L. Hou, L. X. Dai, Tetrahedron: Asymmetry 12(2001) 529-532; (c) B. Jiang, Y. Lei, X. Zhao, J. Org. Chem. 73(2008) 7833-7836; (d) L. Han, Y. Lei, X. Zhao, B. Jiang, J. Org. Chem. 80(2015) 3752-3757; (e) P. J. Pye, K. Rossen, R. A. Reamer, J. Am. Chem. Soc. 119(1997) 6207-6208; (f)C. Bolm, S. Dahmen, C. Bolm, S. Bräse, Angew. Chem. Int. Ed. 41(2002)3692-3694. |
| [14] |
Z. Chen, Y. Huo, P. An, et al., Org. Chem. Front. 3 (2016) 1725-1737. DOI:10.1039/C6QO00386A |
| [15] |
(a) Y. Ma, S. Wei, J. Wu, et al., Adv. Synth. Catal. 350(2008) 2645-2651; (b) H. Lv, Y. Zhang, X. Huang, S. Ye, Adv. Synth. Catal. 350(2008) 2715-2718; (c) L. Zhao, Y. Ma, W. Duan, et al., Org. Lett. 14(2012) 5780-5783. |
| [16] |
(a) J. M. O'Brien, A. H. Hoveyda, J. Am. Chem. Soc. 133(2011) 7712-7715; (b) H. Wu, J. M. Garcia, F. Haeffner, et al., J. Am. Chem. Soc. 137(2015) 10585-10602. |