 2018, Vol. 29
2018, Vol. 29 



b Department of Chemistry, School of Science, Tianjin University, Tianjin 300072, China;
c Institute of New Energy, Shenzhen 518031, China
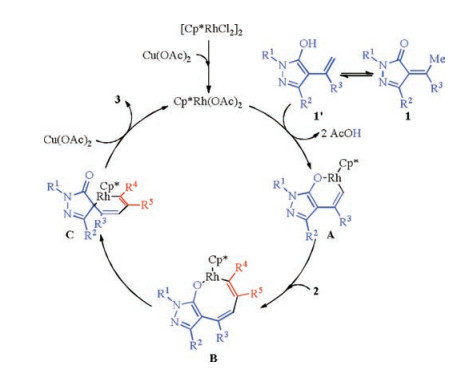
Spirocyclic compounds are common structural motifs in a variety of biologically active natural products, pharmaceutical agents and optoelectronic materials [1]. The development of the efficient methods to synthesis this type of compound has been the subject of the intensive research. In particular, the transition metal-catalyzed C—H bond activation/spiroannulation have recently attracted much research interest [2, 3], which provides one of the most efficient protocols to construct the spirocyclic compounds. In this context, Yao, Lin and co-workers [4, 5] very recently found that with a rhodium complex, the reaction of α-arylidene pyrazolones 1 with alkynes 2 evolves through an formal C(sp3)—H activation/spiroannulation tandem process to selectively generate the spirocyclic product 3 at room temperature (Scheme 1), which provides an efficient protocol for the synthesis of optically active 4-spiro-5-pyrazolones with a five-membered ring. It was suggested that the [RhCp*(OAc)2], generated from the catalyst precursor [RhCp*Cl2]2 with the oxidant Cu(OAc)2, is the active catalytic species (Scheme 2). The substrate 1 first undergoes an isomerization to form dienolate 1', from which the O—H deprotonation and C—H cleavage afford six-membered rhodacycle A. Next, the migratory insertion of alkyne 2 into Rh—C bond generates a rather strained eight-membered rhodacycle B. From intermediate B, an enol-keto tautomerization could occur to deliver the key intermediate C, which could lead to the formation of final product 3 by a subsequent C—C reductive elimination. To be noted, from intermediate B, a C—O reductive elimination could also occur, which could give the experimentally not observed formal [5 + 2] cycloaddition product.

|
Download:
|
| Scheme 1. Rhodium-catalyzed formal C(sp3)—H activation/spiroannulation of α-arylidene pyrazolones 1 with alkynes 2. | |
{kind=link}

|
Download:
|
| Scheme 2. Originally proposed reaction mechanism. | |
{kind=link}
We have recently reported our mechanistic study on the ruthenium- and rhodium-catalyzed oxidative spiroannulation of naphthols and phenols with alkynes [6]. It was found that the originally proposed enol-keto tautomerization/C—C reductive elimination is kinetically inaccessible. Instead, an unusual metallacyclopropene was proposed as a key intermediate to account for the experimental results, from which the C—C coupling could easily lead to the final dearomatized product. You, Zheng, and coworker have reported a computational study on the Rh-catalyzed asymmetric dearomatization of 2-naphthols with alkynes [7]. In that work, the enol-keto tautomerization/C—C reductive elimination was also found to be kinetically unfavorable, and a [1, 3']- reductive elimination directly from the eight-membered rhodacycle intermediate was proposed to account for the formation of the final dearomatized product. Following our continuous research interest in this field [5, 8], we therefore decided to investigate the title reaction by means of density functional theory (DFT) calculations (see the Supporting information for computational details).
In current calculations, (E)-1, 3-diphenyl-4-(1-phenylethylidene)-1H-pyrazol-5(4H)-one 1a and diphenylethyne 2a were selected as the model substrates. The calculated overall energy profile of the rhodium-catalyzed formal C(sp3)—H activation/spiroannulation of 1a with 2a is shown in Fig. 1.

|
Download:
|
| Fig. 1. Calculated energy profile of rhodium-catalyzed formal C(sp3)—H activation/spiroannulation of 1a with 2a. | |
{kind=link}
According to the proposed reaction mechanism (Scheme 2), the reaction begins with the isomerization of (E)-1, 3-diphenyl-4-(1- phenylethylidene)-1H-pyrazol-5(4H)-one 1a to generate dienolate 1a', which was calculated to be endergonic by 7.0 kcal/mol. After the isomerization, intermediate INT1 can be formed through the coordination of 1a' to the Rh center of the active catalyst Cp*Rh(OAc)2, which is slightly more stable than [1a' + Cp*Rh (OAc)2] by 0.3 kcal/mol. From INT1, the O-H deprotonation through transition state TS1 was calculated to be very facile, with an energy barrier of only 4.3 kcal/mol relative to INT1. The O—H deprotonation was found to be exergonic and the resulted intermediate INT2 is 7.2 kcal/mol lower in energy than INT1. Upon the formation of intermediate INT2, the subsequent step is the C(sp2)—H cleavage. This process was found to occur via a six-membered transition state TS2, with an energy barrier of 12.5 kcal/mol relative to INT2. The optimized geometric structure of transition state TS2 shows that it corresponds to the well-established concerted metalation-deprotonation (CMD) mechanism [9], in which one acetate ligand acts as a base to deprotonate the C(sp2)— H with the concomitant formation of the Rh—C(sp2) bond. The intermediate resulted from the C(sp2)—H cleavage is six-membered rhodacycle INT3, which is 1.8 kcal/mol less stable than INT2.
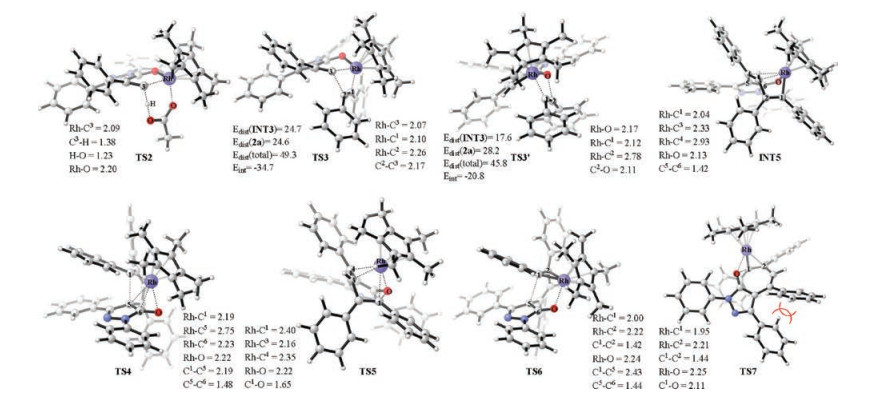
From rhodacycle INT3, the next step is the migratory insertion, starting from the complexation of diphenylethyne 2a with the Rh center to form intermediate INT4. The migratory insertion can take place in two modes, namely the insertion into the Rh—C bond and Rh—O bond. The calculations show that the insertion into the Rh—C bond via transition state TS3 is much more favorable than the insertion into the Rh—O bond via transition state TS3', by as much as 15.3 kcal/mol. To further understand the origins of the energy difference between TS3 and TS3', the distortion/interaction analysis was conducted (Fig. 2) [10]. The results show that the preference of the insertion into the Rh—C bond is mainly determined by the significant difference in the interaction energy. It was found that the overall distortion energy of TS3 is 3.5 kcal/mol higher than that of TS3', while the interaction energy of TS3 is lower than that of TS3' by as much as 13.9 kcal/mol, resulting thus TS3 lower than TS3'. The origins of the significant interaction energy difference could be explained by the comparison of the optimized geometric structures of TS3 and TS3', which shows that in TS3, the Rh center has interactions with both C1 and C2 atoms, while in TS3', no interaction between Rh center and C2 can be found. The insertion into the Rh—C bond was calculated to be highly exergonic and the resulted key eight-membered rhodacycle INT5 is more stable than intermediate INT4 by as much as 24.2 kcal/mol.

|
Download:
|
| Fig. 2. Optimized geometric structures of selected transition states and intermediate; Distances are given in Å. Distortion/interaction energies calculated at the level of B3LYP/6-31G(d, p) (LANL2DZ) are given in kcal/mol. | |
{kind=link}
Upon formation of eight-membered rhodacycle INT5, two possibilities, namely the enol-keto tautomerization/C—C reductive elimination leading to form final spiroannulation product 3a and the C—O reductive elimination leading to form formal [5 + 2] cycloaddition 4a, can be envisioned. The calculations show that the keto-form of INT5 may not exist and several attempts to locate transition state of the C—C reductive elimination were converged to transition state TS4. The IRC calculations show that transition state TS4 is directly connected with the intermediate INT5, instead of the proposed keto-form. From TS4, the catalyst-product complex INT6 can be formed, from which the regeneration of the active catalyst Cp*Rh(OAc)2 with the oxidant Cu(OAc)2 could release final spiroannulation product 3a. In addition, the C—O reductive elimination from INT5 was also considered, which was calculated to take place through transition state TS5. The results show that the C—C reductive elimination via TS4 is only 1.6 kcal/mol lower in energy than the C—O reductive elimination through transition state TS5, which thus not reproduced quite well the experimental results that the spiroannulation product was formed exclusively in the reaction. More importantly, the energy barriers of both possibilities were calculated to be relatively high under the experimental condition (room temperature), being 48.3 and 49.9 kcal/mol relative to INT5.
The calculated results above are indeed consistent with the previous studies on the related ruthenium- and rhodium-catalyzed oxidative spiroannulation of naphthols and phenols with alkynes, in which the proposed enol-keto tautomerization/C—C reductive elimination was also calculated to be kinetically inaccessible [6, 7]. Alternatively, we found that the eight-membered metallacycle has first to isomerize to the unusual metallacyclopropene, from which the C—C coupling could easily lead to the final apiroannulation product [6]. Somewhat different to our work, You, Zheng and coworker proposed that the spiroannulation could be realized by a [1, 3']-reductive elimination directly from the eight-membered metallacycle intermediate [7]. It should be pointed out that the optimized geometric structures of transition states for the C—C coupling and [1, 3']-reductive elimination were found to be quite similar [6, 7]. Based on these previous results, we thus examined the similar possibility in the current reaction.
Indeed, the C—C coupling via transition state TS6 was calculated to be much more favorable than the C—C reductive elimination via transition state TS4, by as much as 24.9 kcal/mol. The optimized geometric structure of TS4 shows that the Rh center has interactions with the C—O bond of the pyrazol ring (Rh—C6 = 2.23 Å, Rh—O = 2.22 Å, Fig. 2), which leads to the interruption of C5—C6 double bond (1.48 Å) [7]. On the other hand in TS6, the bond length of C5—C6 is very close to that in INT5, which may thus leads to the energy of TS6 being much lower than TS4. Although the optimized geometric structure of TS6 displays quite similar to those of the C—C coupling transition states obtained in our previous study [5], the IRC calculations show that TS6 directly connects to eight-membered rhodacycle INT5, instead of the previously proposed metallacyclopropene intermediate. The possible reason that the metallacyclopropene intermediate was not located could be due to that compared with our previous study [6], the presence of the rigid pyrazol ring may cause the similar metallacyclopropene intermediate in the current case being much unstable relative to eight-membered rhodacycle INT5. Additionally, we have previously also found that the formal [5 + 2] cycloaddition can be realized through a novel reductive O—migration [6]. The similar reductive O—migration transition state TS7 was also located in current case, which is 13.5 kcal/mol lower in energy than that of TS5, being in consistent with the experimental outcomes that the spiroannulation product was formed exclusively in the reaction. The origin of the C—C coupling being much more favorable than the reductive O—migration is mainly due to that there is the steric repulsion between two Ph groups in TS7 (Fig. 2), while no such steric repulsion observed in TS6. The same conclusion was also observed in our previous study [6].
In summary, the rhodium-catalyzed formal C(sp3)—H activation/spiroannulation of α-arylidene pyrazolones with alkynes has been investigated by means of DFT calculations. The results show that the reaction is initiated by the keto-enol isomerization of the α-arylidene pyrazolone to form the dienolate intermediate, from which the subsequent O—H deprotonation, C—H cleavage, and migratory insertion afford the eight-membered rhodacycle. The calculations indicate that the spiroannulation through the proposed C—C reductive elimination is kinetically unfeasible. Instead, the C—C coupling from the eight-membered rhodacycle was calculated to be relatively facile, which reproduced quite well the experimental results that the spiroannulation product was formed exclusively in the reaction. The present results collaborate quite well with the previous studies [6, 7], which may provide a general mechanistic scenario for this type of transformation and should be helpful for a better understanding of related reactions.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 21503143), the Tianjin Natural Science Foundation (Nos. 16JCQNJC05600 and 16JCYBJC43600) and the Talent Research Start-up Fund of Tianjin Normal University (No. 5RL139). Yang Cao acknowledges the support from the Shenzhen Peacock Plan (No. 1208040050847074).
| [1] |
(a) For selected reviews, see:, D. A. Horton, G. T. Bourne, M. L. Smythe, Chem. Rev. 103 (2003) 893-930; (b) P. Chauhan, S. Mahajan, D. Enders, Chem. Commun. 51 (2015) 12890-12907. |
| [2] |
(a) M. Gulías, J. L. Mascareñas, Angew Chem. Int. Ed. 55 (2016) 11000-11019; (b) D. L. Davies, S. A. Macgregor, C. L. McMullin, Chem. Rev. 117 (2017) 8649-8709. |
| [3] |
(a) J. Nan, Z. Zuo, L. Luo, et al., J. Am. Chem. Soc. 135 (2013) 17306-17309; (b) J. Zheng, S. B. Wang, C. Zheng, S. L. You, J. Am. Chem. Soc. 137 (2015) 4880-4883; (c) A. Seoane, N. Casanova, N. Quińones, J. L. Mascareńas, M. Gulías, J. Am. Chem. Soc. 136 (2014) 7607-7610; (d) S. Kujawa, D. Best, D. J. Burns, H. W. Lam, Chem. Eur. J. 20 (2014) 8599-8602; (e) Z. Zuo, X. Yang, J. Liu, et al., J. Org. Chem. 80 (2015) 3349-3356; (f) S. Gu, L. Luo, J. Liu, et al., Org. Lett. 16 (2014) 6132-6135; (g) S. R. Chidipudi, I. Khan, H. W. Lam, Angew. Chem. Int. Ed. 51 (2012) 12115-12119; (h) J. D. Dooley, S. R. Chidipudi, H. W. Lam, J. Am. Chem. Soc. 135 (2013) 10829-10836; (i) S. R. Chidipudi, D. J. Burns, I. Khan, H. W. Lam, Angew. Chem. Int. Ed. 54 (2015) 13975-13979. |
| [4] |
J. Zheng, P. Li, M. Gu, A. Lin, H. Yao, Org. Lett. 19 (2017) 2829-2832. DOI:10.1021/acs.orglett.7b00930 |
| [5] |
J. Zheng, S.B. Wang, C. Zheng, S.L. You, Angew. Chem., Int. Ed. 56 (2017) 4540-4544. DOI:10.1002/anie.201700021 |
| [6] |
M. Zhang, G. Huang, Chem. Eur. J. 22 (2016) 9356-9365. DOI:10.1002/chem.201600884 |
| [7] |
C. Zheng, J. Zheng, S.L. You, ACS Catal. 6 (2016) 262-271. DOI:10.1021/acscatal.5b01891 |
| [8] |
(a) G. Huang, P. Liu, ACS Catal. 6 (2016) 809-820; (b) M. Zhang, G. Huang, Dalton Trans. 45 (2016) 3552-3557; (c) G. Huang, J. Org. Chem. 80 (2015) 7564-7571; (d) G. Huang, Org. Lett. 17 (2015) 1994-1997. |
| [9] |
(a) S. R. Neufeldt, G. Jiménez-Osés, J. R. Huckins, O. R. Thiel, K. N. Houk, J. Am. Chem. Soc. 137 (2015) 9843-9854; (b) J. Wu, W. Xu, Z. X. Yu, J. Wang, J. Am. Chem. Soc. 137 (2015) 9489-9496; (c) W. Guo, T. Zhou, Y. Xia, Organometallics 34 (2015) 3012-3020; (d) W. J. Chen, Z. Lin, Organometallics 34 (2015) 309-318; (e) D. L. Davies, C. E. Ellul, S. A. Macgregor, C. L. McMullin, K. Singh, J. Am. Chem. Soc. 137 (2015) 9659-9669; (f) Y. Dang, S. Qu, J. W. Nelson, et al., J. Am. Chem. Soc. 137 (2015) 2006-2014; (g) G. J. Cheng, Y. F. Yang, P. Liu, et al., J. Am. Chem. Soc. 136 (2014) 894-897; (h) M. Anand, R. B. Sunoj, H. F. Schaefer Ⅲ, J. Am. Chem. Soc. 136 (2014) 5535-5538; (i) L. Xu, Q. Zhu, G. Huang, B. Cheng, Y. Xia, J. Org. Chem. 77 (2012) 3017-3204; (j) N. Guimond, S. I. Gorelsky, K. Fagnou, J. Am. Chem. Soc. 133 (2011) 6449-6457; (k) S. Y. Tang, Q. X. Guo, Y. Fu, Chem. -Eur. J. 17 (2011) 13866-13876; (l) S. I. Gorelsky, D. Lapointe, K. Fagnou, J. Am. Chem. Soc. 130 (2008) 10848-10849; (m) D. García-Cuadrado, A. A. C. Braga, F. Maseras, A. M. Echavarren, J. Am. Chem. Soc. 128 (2006) 1066-1067; (n) D. L. Davies, S. M. A. Donald, S. A. Macgregor, J. Am. Chem. Soc. 127 (2005) 13754-13755; (o) B. E. Haines, D. G. Musaev, ACS Catal. 5 (2015) 830-840. |
| [10] |
F.M. Bickelhaupt, K.N. Houk, Angew. Chem. Int. Ed. 56 (2017) 10070-10086. DOI:10.1002/anie.201701486 |