 2018, Vol. 29
2018, Vol. 29 



Chemical synthesis enables precise control on protein composition and incorporation of nonnatural functionality into proteins. Owing to these advantages, chemical synthesis of proteins found wide applications in biochemistry [1], such as protein ubiquitination [2], intein splicing [3], mirror image proteins [4] and all-L and all-D varients of proteins [5]. Meanwhile, chemical synthesis is also an important access to protein pharmaceuticals [6], e.g., nonglycosylated human EPO [7], parathyroid hormone [8] and homogenously glycosylated human interferon-β [9].
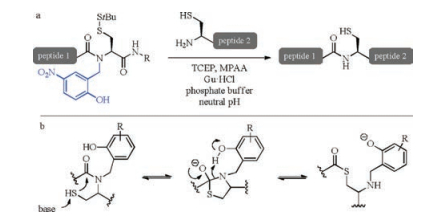
The key to the success of chemical synthesis of proteins is the chemical ligation of unprotected synthetic peptides. Native chemical ligation (NCL), a method developed in 1994 and involving the reactions of C-terminal peptide thioesters and N-terminal cysteine-containing peptide [10], has now evolved into one of the most popular ligation methods [11-13]. Tert-butyloxycarbonyl (Boc) solid phase peptide synthesis (SPPS) approaches were originally used for the preparation of peptide thioester but had been gradually replaced by 9-fluorenylmethoxycarbonyl (Fmoc) SPPS methods due to the troublesome HF treatment [14]. Nevertheless, the piperidine used for the deprotection in Fmoc SPPS approaches also possible wrecks peptide thioesters. To solve this problem, the strategy that peptide thioesters are generated after Fmoc SPPS by treating more stable precursors, e.g., N-acylureas [15, 16], benzotriazole [17] and hydrazides [18, 19] with thiophenol, was developed. On the other hand, intramolecular acyl transfer, which is advantageous in entropy effect, was also utilized to prepare peptide thioester [14]. In the scenario of N-to-S acyl transfer (Scheme 1), amino acids bearing protected mercapto groups are employed for peptide chain elongation. After that, the prepared crypto-thioesters transform into real thioesters via intramolecular N-to-S acyl transfer and react with N-terminal cysteine-containing peptide fragments to complete NCL.

|
Download:
|
| Scheme 1. (a) NCL of N-(2-hydroxybenzyl)cysteine crypto-thioesters and (b) previously proposed mechanism for the corresponding N-to-S acyl transfer. | |
{kind=link}
In this context, various structurally different crypto-thioesters have been developed [20-26], aiming to realize rapid acyl transfer under mild conditions. Very recently, Aucagne and co-workers reported N-(2-hydroxybenzyl)cysteine thioesterification devices (Scheme 1), which rapidly rearrange into thioesters at neutral pH and thus one-pot reactions of thioester formation and NCL are allowed [27-29]. The NCL using Ala and Ser crypto- thioesters can be complete in 24 h at 37 ℃ with the apparent second order kinetic constants close to the values of standard NCL with thioesters [30]. This method was also successfully applied to synthesize the cysteine-rich peptides, and the reduced form of MT7 and CgBigDef1 were obtained with isolated yields of 14% and 18%, respectively. The appealing performance of N-(2-hydroxybenzyl) cysteine thioesterification device was found to result from the phenol substitutes according to control experiments. Specifically, replacing the phenolic hydroxyl by methoxy makes the cryptothioester unreactive. Meanwhile, the acyl transfer/NCL reaction was found to be slightly slower at lower pH than at neutral pH. Blocking the thiol group with stable protecting group leaded to a worse NCL yield, suggesting a N-to-S acyl transfer mechanism rather than a N-to-O shift mechanism. As to the role of the phenol, Aucagne proposed that the amide nitrogen atom is protonated by the phenol via a six-membered transition state during the acyl shift (Scheme 1b).
In addition to experimental studies, the rapid growth of NCL and other peptide ligation methods in recent years has also brought much attention to investigating the relevant reaction mechanisms with theoretical methods [31-39]. However, the detailed mechanism of N-to-S acyl transfer of crypto-thioesters as well as the origin of acceleration effect of substitutes was rarely covered. To consummate the understanding of the thioester formation from crypto-thioesters, a case study on the N-to-S acyl transfer of C-terminal N-(2-hydroxybenzyl)cysteine derivatives was performed in this manuscript with the aid of DFT methods.
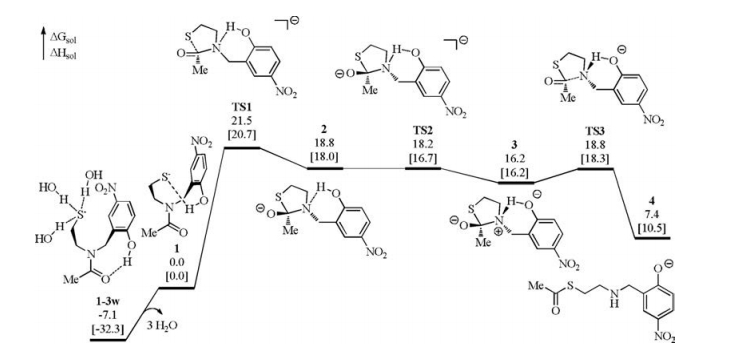
DFT calculations were performed at the level of M06-2x/def2-svp/SMD (solvent = water) (see Supporting information for more detailes) [40-42]. In the presence of tris(2-carboxyethyl)phosphine (TCEP), the disulfide bond can be broken to generate a thiol [43]. As discussed in previous studies [31-37], the thiol further ionizes to generate a thiolate to initiate the N-to-S acyl transfer at neutral pH. Therefore, we chose the thiolate 1 as the model reactant for mechanistic study (Fig. 1). In 1, the phenol moiety forms a hydrogen bond with the thiolate moiety. We examined other conformations in which the phenol forms hydrogen bond with the amide nitrogen atom (1-N) or carbonyl oxygen atom (1-O), or no intramolecular hydrogen bond forms (1-non), but they are less stable than 1 (Scheme S1 in Supporting information). On the other hand, considering the N-to-S acyl transfer proceeds in aqueous solution, extra water was considered to stabilize the thiolate. The interaction of the thiolate with three water molecules (1-3w) is stronger than these with two water molecules (1-2w) or one water molecule (1-1w) (Scheme S1). This is understandable that the thiolate has three lone electron pairs. Although the presence of 1-3w does not disturb the relative feasibility of the different mechanisms discussed after, it is important to explain the observation in the control experiment that changing the hydroxyl to methoxyl (vide infra).

|
Download:
|
| Fig. 1. Calculated energy profile of the N-to-S acyl transfer of 1 starting from the nucleophilic addition of thiolate with the presence of the hydrogen bond between the phenol and the amide nitrogen atom. Solution-phase Gibbs free energies and enthalpies in brackets are given in kcal/mol. | |
{kind=link}
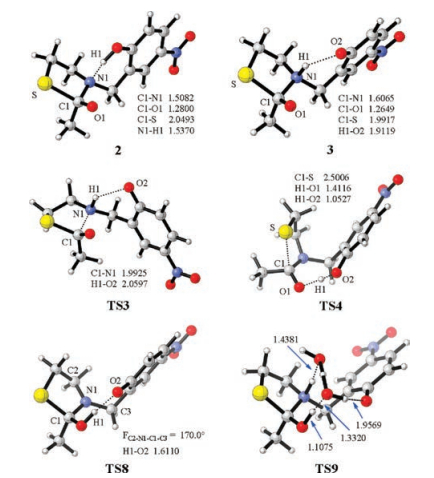
Next, the nucleophilic addition of the thiolate to the carbonyl was investigated. Akin to the Aucagne's proposal (Scheme 1), the phenol was first considered to forms a hydrogen bond with the amide nitrogen to break the conjugation in the amide bond. In this case, the electron-donating effect of the lone electron pair of the nitrogen atom to the carbonyl is expected to be crippled, by which the eletrophilicity of the carbonyl is improved. The corresponding nucleophilic addition occurs via TS1 with an elementary Gibbs free energy barrier of 21.5 kcal/mol referring to 1 (Fig. 1). After TS1, a five-membered-ring alcoholate 2 is formed. Then the direct C—N bond cleavage from 2 to form a thioester was considered but no transition state was located for this process. Relax energy surface scan of the C—N bond from 2 indicates that prior proton transfer is met before the C—N bond cleavage (Fig. S1 in Supporting information). Specifically, the proton transfer from the phenol to the nitrogen atom occurs smoothly via TS2 and generates 3 with an energy decrease of 2.6 kcal/mol. Along with the nitrogen protonation, the C1—N1 bond significantly elongates (1.5082 Å in 2 and 1.6065 Å in 3, Fig. 2) and the C1—O1 bond shortens (1.2800 Å in 2 and 1.2649 Å in 3), suggesting that the proton transfer is conducive to the thioester formation. Indeed, the C—N bond cleavage occurs facilely via TS3 to generate the thioester 4 with a low barrier of 2.6 kcal/mol referring to 3. Note that the N-to-S acyl transfer is endogenic and similar results were also found in previous studies because thioester is less stable than amide [31-39], but the following NCL transforms thioester to amide and is able to drive the reaction forward.

|
Download:
|
| Fig. 2. Structural information of selected intermediates and transition states. Bond lengths are given in Angstrom and dihedral angles are given in degree. | |
{kind=link}
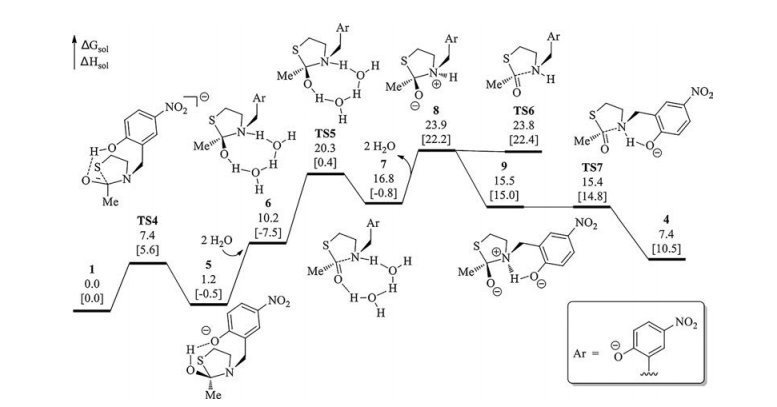
In contrast to TS1, the nucleophilic addition of thiolate to the carbonyl group along with the concerted proton transfer from the phenol to the carbonyl oxygen atom was considered (via TS4, Figs. 2 and 3). It is found that TS4 is much more stable than TS1 by 14.1 kcal/mol although the proton transfer proceeds via an eightmembered-ring transition state. Different to eight-membered rings all consisted by sp3-hybridized carbons, two sp2-hybridized carbon atoms are present in the eight-membered-ring in TS4. Therefore, we speculate that the eight-membered ring for the proton transfer in TS4 cannot be simply considered to have greater ring strain than the six-membered-ring in TS1 based on the knowledge of cyclohexane and cyclooctane. Indeed, 1-non is even more stable than 1-N though an intramolecular hydrogen bond between the phenol and the amide nitrogen atom exist in 1-N (Scheme S1), suggesting that the six-membered ring consisting two sp2-hydridized carbon atoms is not as stable as cyclohexane in the aspect of ring strain. Meanwhile, the protonation of the carbonyl oxygen atom of a model amide Am enlarges the C=O bond by 0.072 Å whereas the protonation of the amide nitrogen atom of Am shortens it by 0.046 Å (Fig. S2 in Supporting information), suggesting that the former is more efficient at breaking the C=O bond to promote the nucleophilic addition. The transformation from 1 to 5 is slightly endergonic by 1.2 kcal/mol whereas the transformation from 1 to 3 causes an energy increase of 16.2 kcal/mol. This result also supports the carbonyl protonation is superior than the amide nitrogen protonation from the aspect of thermodynamics. Akin to 2, the nitrogen protonation is needed for the C—N bond cleavage of 5. For this purpose, water was considered to act as a proton shuttle. 5 combines with two water molecules to generate 6, from which one proton transfer from the carbonyl group to the nitrogen atom formally via TS5 to generate 7 with the aid of water. We also considered that one water molecule assists this process but the water was always captured by the phenolate moiety during geometry optimization thus the corresponding transition state was failed to be located. The departure of the water from 7 generates 8 with an energy increase of 7.1 kcal/mol. 8 can undergoes C—N bond cleavage directly via TS6 or isomerizes to 9 first and then generates thioester via TS7. Because an intramolecular hydrogen bond is present in TS7, TS7 is more stable than TS6 by 8.4 kcal/mol.
In Fig. 1, the initial nucleophilic addition of thiolate is more kinetically difficult than the following steps while the calculated energy profile shown in Fig. 3 has an opposite result. In this case, if one process can connect the initial step of Fig. 3 and the later stage of Fig. 1, a lower overall energy barrier is expected. Inspiring by our previous study which involves the inversion of amines [39], the isomerization of 5 to 10 via TS8 was located (Fig. 4). This process is allowed with an energy barrier of 4.2 kcal/mol referring to 5. Thereafter, water-assisted proton transfer is realized via TS9 and generates 12, which further undergoes C—N bond cleavage via TS10 to give 4. By comparing the calculated energy profiles in Figs. 1, 3 and 4, the most feasible pathways among candidates considered involves the four major steps: the concerted nucleophilic addition of thiolate/proton transfer via TS4, the inversion of the amine moiety via TS8, the water-assisted proton transfer via TS9 and the C—N bond cleavage via TS10. The rate-determining step is TS9 with an overall Gibbs free energy barrier of 20.4 kcal/mol referring to 1-3w, which is well consistent with the experimental conditions of 24 h at 37 ℃.

|
Download:
|
| Fig. 3. Calculated energy profile of the N-to-S acyl transfer of 1 starting by concerted nucleophilic addition of thiolate/proton transfer. Solution-phase Gibbs free energies and enthalpies in brackets are given in kcal/mol. | |
{kind=link}

|
Download:
|
| Fig. 4. Calculated energy profile of the inversion of the amine moiety on 5 and the subsequent steps of N-to-S acyl transfer. Solution-phase Gibbs free energies and enthalpies are given in kcal/mol. | |
{kind=link}
To further check the validity of our proposed mechanism, the energy profile of the reported control experiment where the hydroxyl was changed to methoxyl (1a), was calculated for comparison (Fig. 5). It was found that the energy difference between 1a and 1-3wa is greater than that between 1 and 1-3w. This is understandable that no intramolecular hydrogen bond is present to stabilize the 1a. By contrast, the energy difference between TS1a and 1a is smaller than that between TS1 and 1. Because no intramolecular hydrogen bond is present in TS1a and 1a while one hydrogen bond is present in both TS1 and 1, it seems that the interaction between the phenol and the thiolate provides stronger stabilization effect than that between the phenol and the carbonyl oxygen. The relative stabilities of 1-N and 1-O also supports this speculation (Scheme S1). However, 1a experiences a more kinetically difficult thiolate nucleophilic addition with an overall Gibbs free energy barrier of 31.5 kcal/mol. Comparing the process 1-3wa→TS1a with the process of 1-3w→TS1, an intramolecular hydrogen bond exists and activates the carbonyl in the latter case whereas no such extra factor exists in the former case to promote the nucleophilic addition. The direct C—N bond cleavage from anionic 2a can occur via TS3a' but the corresponding overall Gibbs free energy barrier exceeds 60 kcal/mol, and thus we excluded this pathway. Referring to the discussion about TS2, the protonation of the nitrogen atom was considered before the C—N bond cleavage. Due to the absence of intramolecular proton donor in the acyl transfer of 1a, the protonation of 2a by an extra protonated water to generate 3a was calculated. Different to the process of 2→3, the transformation of 2a–3a involves the combination of a cation (H3O+) and an anion (2a). Therefore, we proposed that the corresponding strong electrostatic interaction is responsible for the remarkable energy decrease of 34.8 kcal/mol. From 3a, rapid C—N bond cleavage occurs via TS3a to complete the N-to-S acyl transfer with a Gibbs free energy barrier of 1.6 kcal/mol, which is very close to the energetic difference between 3 and TS3. This result indicates that the phenolate and the methoxyl differ little in the final C—N bond cleavage step. According to the energy profile shown in Fig. 5, the nucleophilic addition of thiolate is the rate-determining step in the N-to-S acyl transfer of 1a, with an overall energy barrier of 31.5 kcal/mol, a value that is higher than that of 1 by 10.1 kcal/mol. This result is qualitatively consistent with the experimental observation that changing the phenolic hydroxyl to methoxyl makes the crypto-thioester much less reactive, and thus further gives a support for our study.

|
Download:
|
| Fig. 5. Calculated energy profile of the N-to-S acyl transfer of 1a. Solution-phase Gibbs free energies and enthalpies are given in kcal/mol. | |
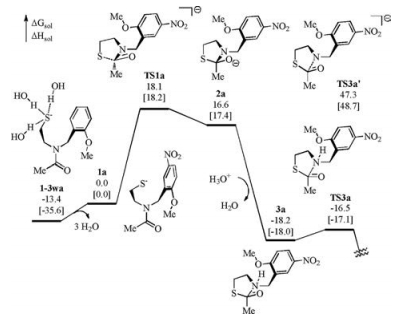{kind=link}
In conclusion, N-(2-hydroxybenzyl)cysteine derivatives are efficient crypto-thioesters that can be directly employed for NCL at neutral pH with fast kinetics. To elucidate the mechanism of the related N-to-S acyl transfer and the origin of the acceleration effect of the phenol substitutes, a DFT study was performed in this manuscript. It was found that the N-to-S acyl transfer of N-(2-hydroxybenzyl)cysteine derivatives proceeds via the concerted nucleophilic addition of thiolate/proton transfer, the inversion of the amine moiety, the water-assisted proton transfer to protonate the amide nitrogen atom and the C—N bond cleavage. With the presence of the phenol substitutes, the nucleophilic addition of thiolate is significantly accelerated due to the hydrogen bond forming between the phenolic hydroxyl group and the carbonyl oxygen. In this case, the water-assisted proton transfer from protonated carbonyl to the amide nitrogen atom is the ratedetermining step of the N-to-S acyl transfer. By contrast, changing the phenolic hydroxyl to methoxyl was found to hinder the nucleophilic addition of thiolate and the relevant N-to-S acyl transfer becomes slower accordingly. This computational result agrees with the reported control experiments and gives a support for our study. Compared with the previous mechanistic studies on N-to-S acyl transfer, this manuscript provides a rare example on the N-to-S acyl transfer assisted by intramolecular substitutes and clarified the self-catalytic role of the substitutes. We hope that the presented results help understanding the N-to-S acyl transfer of other crypto-thioesters and inspire the further experimental design.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21702119, 21473100, 21603116 and 21703118), Natural Science Foundation of Shandong Province (Nos. ZR2017QB001, ZR2017MB038), Special Fund Project for Postdoctoral Innovation of Shandong Province (No. 201602021).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2017.11.043.
| [1] |
S.B.H. Kent, Chem. Soc. Rev. 38 (2009) 338-351. DOI:10.1039/B700141J |
| [2] |
M. Pan, S. Gao, Y. Zheng, et al., J. Am. Chem. Soc. 138 (2016) 7429-7435. DOI:10.1021/jacs.6b04031 |
| [3] |
J. Binschik, H.D. Mootz, Angew. Chem. Int. Ed. 52 (2013) 4260-4264. DOI:10.1002/anie.201208863 |
| [4] |
Z. Wang, W. Xu, L. Liu, T.F. Zhu, Nat. Chem. 8 (2016) 698-704. DOI:10.1038/nchem.2517 |
| [5] |
A.M. Levinson, J.H. McGee, A.G. Roberts, et al., J. Am. Chem. Soc. 139 (2017) 7632-7639. DOI:10.1021/jacs.7b02988 |
| [6] |
J.M. Chalker, Chem. Biol. Drug Des. 81 (2013) 122-135. DOI:10.1111/cbdd.2012.81.issue-1 |
| [7] |
S. Liu, B.L. Pentelute, S.B.H. Kent, Angew. Chem. Int. Ed. 51 (2012) 993-999. DOI:10.1002/anie.v51.4 |
| [8] |
S. Shang, Z. Tan, S.J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 5986-5989. DOI:10.1073/pnas.1103118108 |
| [9] |
I. Sakamoto, K. Tezuka, K. Fukae, et al., J. Am. Chem. Soc. 134 (2012) 5428-5431. DOI:10.1021/ja2109079 |
| [10] |
P.E. Dawson, T.W. Muir, I. Clark-Lewis, S.B. Kent, Science 266 (1994) 776-779. DOI:10.1126/science.7973629 |
| [11] |
Y.C. Huang, G.M. Fang, L. Liu, Natl. Sci. Rev. 3 (2016) 107-116. DOI:10.1093/nsr/nwv072 |
| [12] |
S. Bondalapati, M. Jbara, A. Brik, Nat. Chem. 8 (2016) 407-418. DOI:10.1038/nchem.2476 |
| [13] |
O. Koniev, A. Wagner, Chem. Soc. Rev. 44 (2015) 5495-5551. DOI:10.1039/C5CS00048C |
| [14] |
H. Li, S. Dong, Sci. China Chem. 60 (2017) 201-213. |
| [15] |
J.B. Blanco-Canosa, P.E. Dawson, Angew. Chem. Int. Ed. 47 (2008) 6851-6855. DOI:10.1002/anie.v47:36 |
| [16] |
J.B. Blanco-Canosa, B. Nardone, F. Albericio, P.E. Dawson, J. Am. Chem. Soc. 137 (2015) 7197-7209. DOI:10.1021/jacs.5b03504 |
| [17] |
J.X. Wang, G.M. Fang, Y. He, et al., Angew. Chem. Int. Ed. 54 (2015) 2194-2198. DOI:10.1002/anie.201408078 |
| [18] |
G.M. Fang, Y.M. Li, F. Shen, et al., Angew. Chem. Int. Ed. 50 (2011) 7645-7649. DOI:10.1002/anie.201100996 |
| [19] |
G.M. Fang, J.X. Wang, L. Liu, Angew. Chem. Int. Ed. 51 (2012) 10347-10350. DOI:10.1002/anie.201203843 |
| [20] |
T. Kawakami, Top. Curr. Chem. 362 (2015) 107-135. |
| [21] |
J.S. Zheng, H.N. Chang, F.L. Wang, L. Liu, J. Am. Chem. Soc. 133 (2011) 11080-11083. DOI:10.1021/ja204088a |
| [22] |
L. Raibaut, M. Cargoët, N. Ollivier, et al., Chem. Sci. 7 (2016) 2657-2665. DOI:10.1039/C5SC03459K |
| [23] |
S.L. Pira, O. El Mahdi, L. Raibaut, et al., Org. Biomol. Chem. 14 (2016) 7211-7216. DOI:10.1039/C6OB01079B |
| [24] |
C. Rao, C.F. Liu, Org. Biomol. Chem. 15 (2017) 2491-2496. DOI:10.1039/C7OB00103G |
| [25] |
H.E. Elashal, Y.E. Sim, M. Raj, Chem. Sci. 8 (2017) 117-123. DOI:10.1039/C6SC02162J |
| [26] |
S. Tsuda, M. Mochizuki, K. Sakamoto, et al., Org. Lett. 18 (2016) 5940-5943. DOI:10.1021/acs.orglett.6b03055 |
| [27] |
V.P. Terrier, H. Adihou, M. Arnould, A.F. Delmas, V. Aucagne, Chem. Sci. 7 (2016) 339-345. DOI:10.1039/C5SC02630J |
| [28] |
D. Lelievre, V.P. Terrier, A.F. Delmas, V. Aucagne, Org. Lett. 18 (2016) 920-923. DOI:10.1021/acs.orglett.5b03612 |
| [29] |
V.P. Terrier, A.F. Delmas, V. Aucagne, Org. Biomol. Chem. 15 (2017) 316-319. DOI:10.1039/C6OB02546C |
| [30] |
S.B. Pollock, S.B.H. Kent, Chem. Commun. 47 (2011) 2342-2344. DOI:10.1039/C0CC04120C |
| [31] |
C. Wang, Q.X. Guo, Y. Fu, Chem.-Asian J. 6 (2011) 1241-1251. DOI:10.1002/asia.201000760 |
| [32] |
C. Wang, L. Liu, Chin. J. Chem. 30 (2012) 1974-1979. DOI:10.1002/cjoc.201200337 |
| [33] |
C. Wang, Q.X. Guo, Sci. China Chem. 55 (2012) 2075-2080. DOI:10.1007/s11426-012-4711-x |
| [34] |
Q. Zhang, H.Z. Yu, J. Shi, Acta Phys. Chim. Sin. 29 (2013) 2321-2331. |
| [35] |
X.H. Sun, H.Z. Yu, M.M. Yang, Y.M. Yang, Z.M. Dang, J. Phys. Org. Chem. 28 (2015) 586-590. DOI:10.1002/poc.v28.9 |
| [36] |
X.H. Sun, H.Z. Yu, S.Q. Pei, Z.M. Dang, Chin. Chem. Lett. 26 (2015) 1259-1264. DOI:10.1016/j.cclet.2015.07.003 |
| [37] |
M.I. Ali Shah, Z.Y. Xu, L. Liu, Y.Y. Jiang, J. Shi, RSC Adv. 6 (2016) 68312-68321. DOI:10.1039/C6RA13793H |
| [38] |
Y.Y. Jiang, C. Wang, Y. Liang, et al., J. Org. Chem. 82 (2017) 1064-1072. DOI:10.1021/acs.joc.6b02642 |
| [39] |
Y.Y. Jiang, L. Zhu, X. Man, Y. Liang, S. Bi, Tetrahedron 73 (2017) 4380-4386. DOI:10.1016/j.tet.2017.05.099 |
| [40] |
Y. Zhao, D.G. Truhlar, Theor. Chem. Acc. 120 (2008) 215-241. DOI:10.1007/s00214-007-0310-x |
| [41] |
A.V. Marenich, C.J. Cramer, D.G. Truhlar, J. Phys. Chem. B 113 (2009) 6378-6396. DOI:10.1021/jp810292n |
| [42] |
F. Weigend, R. Ahlrichs, Phys. Chem. Chem. Phys. 7 (2005) 3297-3305. DOI:10.1039/b508541a |
| [43] |
U.T. Rüegg, J. Rudinger, Methods Enzymol. 47 (1977) 111-116. DOI:10.1016/0076-6879(77)47012-5 |