 2018, Vol. 29
2018, Vol. 29 



b College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450001, China
The development of organocatalysis [1-13], which avoid the use of transition metals, is of great interest because of the extensive applications of these species in organic synthesis [14-24]. Among various organocatalysts, phosphoric acids catalysis [25-32] has become a powerful tool for the construction of C—C (X) bonds in the synthesis of carbocyclic, functionalized spirocyclic, and fused-ring systems, which are widely used in the synthesis of bioactive natural products and medicinally important substances. Generally, phosphoric acids are organic acids, and are often molecularly dissolved in organic solvents. The -OH moiety of a phosphoric acid is expected to capture electrophilic organic components through hydrogen-bonding interactions, resulting in lowest unoccupied molecular orbital (LUMO)-lowering activity [33-37]. The phosphoryl oxygen can also function as a Brønsted basic site to accept a proton [38-42]. Phosphoric acids are therefore anticipated to be acid/base dual function catalysts.
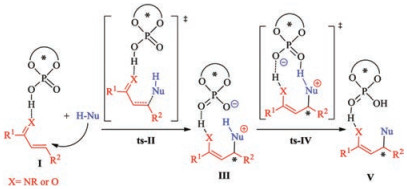
Given the utility of phosphoric acids, the scope of phosphoric acid-catalyzed reactions, especially asymmetric transformations [43-49], has expanded greatly. Chiral phosphoric acids are excellent catalysts for Friedel–Crafts-type reactions of nucleophiles with activated olefins such as α, β-unsaturated ketones, nitroolefins, and α, β-unsaturated imines [50-55]. The rapid progress in experimental aspects of these reactions has made mechanistic studies of this type of reaction an urgent issue, especially clarification of the origin of the stereoselectivity [56-63]. The generally accepted mechanism starts with hydrogenbonding activation of a catalyst with an electrophilic component (Scheme 1). Nucleophilic addition to the activated olefin moiety affords a stereocenter via transition state ts-Ⅱ. Subsequent phosphoric acid-assisted proton transfer via transition state ts-Ⅳ leads to generation of the product. In previous investigations, nucleophilic addition was often considered to be the ratedetermining step as well as the stereoselectivity-determining step [64-68]. However, nucleophilic addition is usually an endergonic process, which suggests that the energy barrier to the subsequent deprotonation would not be negligible, especially in the reaction between an inert C—H bond and a nucleophile. Taking these into consideration, a thorough theoretical investigation of the mechanism and origin of the stereoselectivity of phosphoric acid-catalyzed transformations is urgently and indispensable.

|
Download:
|
| Scheme 1. Proposed mechanism for phosphoric acid-catalyzed Friedel–Crafts-type reactions. | |
{kind=link}
Recently, List and Coelho et al. [69] jointly succeeded in developing a phosphoric acid-catalyzed asymmetric conjugate addition of indolizines to α, β-unsaturated ketones (Scheme 2). This phosphoric acid-catalyzed Friedel–Crafts-type reaction has good functional group tolerance, a broad substrate scope, and high enantioselectivity (up to 98:2). Because of the significance of this transformation, it is necessary to elucidate the reaction mechanism, including the origins of the observed enantioselectivity. Here, we performed density functional theory (DFT) calculations to investigate this reaction.

|
Download:
|
| Scheme 2. Phosphoric acid-catalyzed asymmetric conjugate addition of indolizines to α, β-unsaturated ketones. | |
{kind=link}
All calculations were performed with the Gaussian 09 program [70]. Geometry optimization of the minimum energy structures and transition states was performed at the B3-LYP [71, 72] level of theory with the 6-31G(d) basis set. Harmonic vibrational frequency calculations were performed for all stationary points to determine whether they were local minima or transition structures, and to derive the thermochemical corrections for the enthalpies and free energies. Solvent effects in benzene were considered implicitly by performing single-point energy calculations on the gas-phase optimized geometries, using the SMD polarizable continuum model [73-78]. Solvation single-point energies were obtained using the M06 [79-83] functional with the 6–311 + G(d, p) basis set, in which dispersion interactions were included. In our computational studies, non-covalent interactions (NCI) analyses of key transition states were obtained with the Multiwfn [84-87].
Based on previous theoretical and experimental studies, the proposed reaction pathways for the phosphoric acid-catalyzed asymmetric conjugate addition of indolizines to α, β-unsaturated ketones are shown in Scheme 3. Initially, molecular phosphoric acid captures a conjugated ketone component through hydrogenbonding interactions with an -OH moiety, which results in activation of the conjugated ketone by lowering the LUMO. Nucleophilic addition of an indolizine to the activated olefin moiety leads to formation of a new C—C bond, and two chiral centers are constructed in intermediate C. Subsequent deprotonation–aromatization affords the enol intermediate D, which tautomerizes to the asymmetric ketone product E. However, the following aspects of the proposed mechanism remain unclear.(1) Which step (nucleophilic addition or deprotonation–aromatization) should be considered to be the rate-determining step and enantioselectivity-determining step? (2) What is the origin of the enantioselectivity?

|
Download:
|
| Scheme 3. Proposed mechanism for phosphoric acid-catalyzed asymmetric conjugate addition of indolizines to α, β-unsaturated ketones. | |
{kind=link}
Fig. 1 shows that the hydrogen-bonding interactions between phosphoric acid catalyst 3 and conjugated ketone 2 afford the active intermediate 5, with a free-energy decrease of 1.7 kcal/mol. Subsequent nucleophilic addition of indolizine 1 to the re face of the activated olefin occurs via transition state 6-ts-SR, with a freeenergy barrier of 25.4 kcal/mol, leading to reversible generation of indolizinium intermediate 7-SR. Deprotonation–aromatization then generates enol intermediate 9-S via transition state 8-tsSR, which has a barrier of only 6.1 kcal/mol. However, the relative free energy of transition state 8-ts-SR is 0.1 kcal/mol higher than that of 6-ts-SR because of the endergonic nucleophilic addition process. This suggests that the deprotonation–aromatization step is the rate-determining step in this catalytic cycle. The subsequent tautomerization exothermically releases the asymmetric ketone product 4-S, with concurrent regeneration of the phosphoric acid catalyst 3.

|
Download:
|
| Fig. 1. Free energy profiles for phosphoric acid-catalyzed asymmetric conjugate addition of indolizines to α, β-unsaturated ketones (R = CO2Me). The values given by kcal/mol are the relative free energies calculated by M06 method in benzene solvent. | |
{kind=link}
Another reaction pathway, which leads to the enantiomer 4-R, was also calculated, and is shown in Fig. 1. Starting from intermediate 5, nucleophilic addition of indolizine 1 to the si face of the activated olefin via transition state 6-ts-RR could afford indolizinium 7-RR with the (R, R) configuration. The corresponding deprotonation–aromatization then generates enol intermediate 9-R via transition state 8-ts-RR. The relative free energy of 8-ts-RR is 6.6 kcal/mol higher than that of 6-ts-RR, which clearly indicates that the deprotonation–aromatization step should be considered as the rate-determining step in the catalytic cycle. In addition, we considered two other competitive pathways involving (R, S)-or (S, S)-configured indolizinium intermediates (7-RS or 7-SS), leading to products 4-R or 4-S, respectively. The detailed free-energy profiles are shown in Fig. S1 in Supporting information. The corresponding overall activation free energies of the ratedetermining steps for these two pathways are higher than those shown in Fig. 1.
It is also worth noting that rotation of the C—C single bond in the conjugated ketone could provide an s-trans isomer. As shown in Fig. 2, this rotation proceeds via transition state 10-ts, with a free- energy barrier of 9.5 kcal/mol, leading to generation of the s-trans isomer 11. The relative free energy of intermediate 11 is only 0.4 kcal/mol higher than that of its s-cis isomer 5. This indicates that there is a rapid thermodynamic equilibrium between these two isomers. Accordingly, the analogous reaction pathways leading to products 4-R or 4-S were also considered, with intermediate 11 as the starting material. The nucleophilic addition of indolizine 1 to the re face of the activated olefin via 12-ts-SR generates the intermediate 13-SR. Subsequent deprotonation– aromatization, followed by tautomerization, affords the product 4-S. The overall activation free energy of this pathway is 8.7 kcal/mol higher than that of the corresponding pathway for 4-S generation shown in Fig. 1. This suggests that 4-S is preferentially generated via the pathway starting from the cis intermediate 5. In the alternative pathway, nucleophilic addition on the si face of the strans activated olefin 11 could generate product 4-R. However, the overall activation free energy of this pathway is 27.5 kcal/mol, which is 42.3 kcal/mol lower than that for the pathway for 4-R generation shown in Fig. 1. The calculated energy discrepancy shows that the generation of side product 4-R preferentially occurs via the pathway starting from 11.

|
Download:
|
| Fig. 2. Free energy profiles for competitive pathways for the competitive pathways via trans conjugated ketone (R = CO2Me). The values given by kcal/mol are the relative free energies calculated by M06 method in benzene solvent. | |
{kind=link}
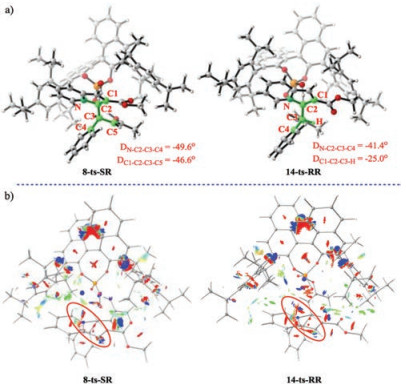
As shown in Figs. 1 and 2, products 4-S and 4-R are considered to be generated via the reaction pathways starting from s-cis intermediate 5 and s-trans intermediate 11, respectively. Our theoretical calculations show that deprotonation–aromatization is the rate-determining and enantioselectivity-determining step. The relative free energy of 8-ts-SR is 2.0 kcal/mol lower than that of 14-ts-RR, indicating that product 4-S should be the major product obtained experimentally with phosphoric acid 3. The theoretical study predicts that the ee value of product 4-S is 93%, which is consistent with experimental value (91%). Optimized structure analysis (Fig. 3a) shows that in transition state 8-ts-SR, the dihedral angles DN—C2–C3–C4 and DC1–C2–C3–C5 are —49.6° and —46.6°, respectively. However, the corresponding dihedral angles DN—C2–C3–C4 and DC1–C2–C3–H in transition state 14-ts-RR are only —41.4° and —25.0°, respectively. These data show that the C2—C3 bond could not adopt a completely staggered conformation in transition state 14-ts-RR driven by the steric hindrance between the reactant and catalyst pocket. The bond overlap strain results in a higher free energy for transition state 14-ts-RR. The intrinsic effects of enantioselectivity were evaluated by performing NCI analysis for 8-ts-SR and 14-ts-RR. The results show that the energetic discrepancy was mainly caused by the significant steric effect in 14-ts-RR.

|
Download:
|
| Fig. 3. (a) Optimized structures of transition state 8-ts-SR and 14-ts-RR. (b) Noncovalent interaction (NCI) analysis for 8-ts-SR and 14-ts-RR. (blue, strong attraction; green, weak attractive interaction; red, steric effect.). | |
{kind=link}
In conclusion, DFT calculations were used to investigate the mechanism and enantioselectivity of phosphoric acid-catalyzed asymmetric conjugate addition of indolizines to α, β-unsaturated ketones. The calculation results showed that this transformation occurs via a reaction pathway involving nucleophilic addition from the indolizine to the hydrogen-bond-activated olefin, deprotonation–aromatization, and tautomerization. Deprotonation–aromatization is considered to be the ratedetermining and enantioselectivity-determining step. The product with S configuration is preferentially generated via the pathway starting from an s-cis conjugated ketone, whereas the product with R configuration is preferentially generated via the pathway starting from the corresponding s-trans isomer. Further optimized structure and NCI analyses showed that the main origin of the enantioselectivity is bond-rotation strain in the transition states of the deprotonation–aromatization step.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 21772020). We are also grateful to the Fundamental Research Funds for the Central Universities (Chongqing University) (No. 106112017CDJXY220007).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.03.018.
| [1] |
J. Seayad, B. List, Org. Biomol. Chem. 3 (2005) 719-724. DOI:10.1039/b415217b |
| [2] |
D.W.C. MacMillan, Nature 455 (2008) 304-308. DOI:10.1038/nature07367 |
| [3] |
P. Li, W.H. Wang, S.W. Bi, R. Song, Y.X. Bu, Sci. China Ser. B 52 (2009) 131-136. DOI:10.1007/s11426-009-0022-2 |
| [4] |
J. Xiao, K. Zhao, T.P. Loh, Chem. Commun. 48 (2012) 3548-3550. DOI:10.1039/c2cc30261f |
| [5] |
Q. Zhang, H.Z. Yu, Y. Fu, Org. Chem. Front. 1 (2014) 614-624. DOI:10.1039/C4QO00036F |
| [6] |
T. Wang, Z. Yu, D.L. Hoon, et al., Chem. Sci. 6 (2015) 4912-4922. DOI:10.1039/C5SC01614B |
| [7] |
H. Zhao, M. Xiao, L. Xu, L. Wang, J. Xiao, RSC Adv. 6 (2016) 38558-38562. DOI:10.1039/C5RA21614A |
| [8] |
C. Min, D. Seidel, Chem. Soc. Rev. 46 (2017) 5889-5902. DOI:10.1039/C6CS00239K |
| [9] |
B. Wang, T. Xu, L. Zhu, et al., Org. Chem. Front. 4 (2017) 1266-1271. DOI:10.1039/C7QO00124J |
| [10] |
B. Wu, Z. Yu, X. Gao, Y. Lan, Y.G. Zhou, Angew. Chem. Int. Ed. 56 (2017) 4006-4010. DOI:10.1002/anie.201700437 |
| [11] |
Q.F. Cheng, J.W. Wang, Q.F. Wang, Z. Liu, Chin. Chem. Lett. 27 (2016) 1032-1035. DOI:10.1016/j.cclet.2016.03.007 |
| [12] |
Y.N. Gao, M. Shi, Chin. Chem. Lett. 28 (2017) 493-502. DOI:10.1016/j.cclet.2016.12.001 |
| [13] |
Z.Z. Yuan, X.W. Kong, L.H. Liu, H.X. Zhu, H. Xiao, Chin. Chem. Lett. 28 (2017) 1469-1472. DOI:10.1016/j.cclet.2017.02.017 |
| [14] |
R. Dalpozzo, G. Bartoli, G. Bencivenni, Chem. Soc. Rev. 41 (2012) 7247-7290. DOI:10.1039/c2cs35100e |
| [15] |
X. Jiang, X. Shi, S. Wang, et al., Angew. Chem. Int. Ed. 51 (2012) 2084-2087. DOI:10.1002/anie.201107716 |
| [16] |
M.Y. Han, J.Y. Jia, W. Wang, Tetrahedron Lett. 55 (2014) 784-794. DOI:10.1016/j.tetlet.2013.11.048 |
| [17] |
D.B. Ramachary, A.B. Shashank, S. Karthik, Angew. Chem. Int. Ed. 53 (2014) 10420-10424. DOI:10.1002/anie.201406721 |
| [18] |
H. Gao, Q.L. Xu, C. Keene, et al., Angew. Chem. Int. Ed. 55 (2016) 566-571. DOI:10.1002/anie.201508419 |
| [19] |
W. Yao, Z. Yu, S. Wen, et al., Chem. Sci. 8 (2017) 5196-5200. DOI:10.1039/C7SC00952F |
| [20] |
S.S. Li, L. Zhou, L. Wang, et al., Org. Lett. 20 (2018) 138-141. DOI:10.1021/acs.orglett.7b03492 |
| [21] |
P.S. Reddy, M.V.K. Reddy, P.V.G. Reddy, Chin. Chem. Lett. 27 (2016) 943-947. DOI:10.1016/j.cclet.2016.01.046 |
| [22] |
Z.K. Xue, N.K. Fu, S.Z. Luo, Chin. Chem. Lett. 28 (2017) 1083-1086. DOI:10.1016/j.cclet.2017.01.014 |
| [23] |
X.D. Zhai, Z.D. Yang, Z. Luo, H.T. Xu, Chin. Chem. Lett. 28 (2017) 1793-1797. DOI:10.1016/j.cclet.2017.04.017 |
| [24] |
K. Sun, X.L. Chen, X. Li, et al., Chem. Commun. 51 (2015) 12111-12114. DOI:10.1039/C5CC04484G |
| [25] |
G.B. Rowland, E.B. Rowland, Y. Liang, J.A. Perman, J.C. Antilla, Org. Lett. 9 (2007) 2609-2611. DOI:10.1021/ol0703579 |
| [26] |
Y. He, M. Lin, Z. Li, et al., Org. Lett. 13 (2011) 4490-4493. DOI:10.1021/ol2018328 |
| [27] |
J. Lv, S. Luo, Chem. Commun. 49 (2013) 847-858. DOI:10.1039/C2CC34288J |
| [28] |
Y.C. Zhang, J.J. Zhao, F. Jiang, S.B. Sun, F. Shi, Angew. Chem. Int. Ed. 53 (2014) 13912-13915. DOI:10.1002/anie.201408551 |
| [29] |
F.E. Held, D. Grau, S.B. Tsogoeva, Molecules 20 (2015) 16103-16126. DOI:10.3390/molecules200916103 |
| [30] |
X. Yang, F.D. Toste, J. Am. Chem. Soc. 137 (2015) 3205-3208. DOI:10.1021/jacs.5b00229 |
| [31] |
M.N. Grayson, Z. Yang, K.N. Houk, J. Am. Chem. Soc. 139 (2017) 7717-7720. DOI:10.1021/jacs.7b03847 |
| [32] |
Y. Shimoda, H. Yamamoto, J. Am. Chem. Soc. 139 (2017) 6855-6858. DOI:10.1021/jacs.7b03592 |
| [33] |
M. Terada, Synthesis (2010) 1929-1982. |
| [34] |
K. Brak, E.N. Jacobsen, Angew. Chem. Int. Ed. 52 (2013) 534-561. DOI:10.1002/anie.201205449 |
| [35] |
Z. Wang, F.K. Sheong, H.H. Sung, et al., J. Am. Chem. Soc. 137 (2015) 5895-5898. DOI:10.1021/jacs.5b03083 |
| [36] |
M.R. Monaco, D. Fazzi, N. Tsuji, et al., J. Am. Chem. Soc. 138 (2016) 14740-14749. DOI:10.1021/jacs.6b09179 |
| [37] |
L. Bayeh, P.Q. Le, U.K. Tambar, Nature 547 (2017) 196-200. DOI:10.1038/nature22805 |
| [38] |
S.J. Connon, Angew. Chem. Int. Ed. 45 (2006) 3909-3912. DOI:10.1002/(ISSN)1521-3773 |
| [39] |
M. Terada, Chem. Commun. (2008) 4097-4112. |
| [40] |
M. Hatano, K. Moriyama, T. Maki, K. Ishihara, Angew. Chem. Int. Ed. 49 (2010) 3823-3826. DOI:10.1002/anie.v49:22 |
| [41] |
I. Coric, B. List, Nature 483 (2012) 315-319. DOI:10.1038/nature10932 |
| [42] |
H.J. Pan, Y. Zhang, C. Shan, et al., Angew. Chem. Int. Ed. 55 (2016) 9615-9619. DOI:10.1002/anie.201604025 |
| [43] |
S. Hoffmann, A.M. Seayad, B. List, Angew. Chem. Int. Ed. 117 (2005) 7590-7593. DOI:10.1002/(ISSN)1521-3757 |
| [44] |
S. Xu, Z. Wang, X. Zhang, X. Zhang, K. Ding, Angew. Chem. Int. Ed. 120 (2008) 2882-2885. DOI:10.1002/(ISSN)1521-3757 |
| [45] |
D. Parmar, E. Sugiono, S. Raja, M. Rueping, Chem. Rev. 114 (2014) 9047-9153. DOI:10.1021/cr5001496 |
| [46] |
X. Tian, N. Hofmann, P. Melchiorre, Angew. Chem. Int. Ed. 53 (2014) 2997-3000. DOI:10.1002/anie.v53.11 |
| [47] |
N. Dong, Z.P. Zhang, X.S. Xue, X. Li, J.P. Cheng, Angew. Chem. Int. Ed. 55 (2016) 1460-1464. DOI:10.1002/anie.201509110 |
| [48] |
S. Li, J.W. Zhang, X.L. Li, D.J. Cheng, B. Tan, J. Am. Chem. Soc. 138 (2016) 16561-16566. DOI:10.1021/jacs.6b11435 |
| [49] |
J.P. Reid, L. Simon, J.M. Goodman, Acc. Chem. Res. 49 (2016) 1029-1041. DOI:10.1021/acs.accounts.6b00052 |
| [50] |
Y.X. Jia, J. Zhong, S.F. Zhu, C.M. Zhang, Q.L. Zhou, Angew. Chem. Int. Ed. 46 (2007) 5565-5567. DOI:10.1002/(ISSN)1521-3773 |
| [51] |
Q. Kang, Z.A. Zhao, S.L. You, J. Am. Chem. Soc. 129 (2007) 1484-1485. DOI:10.1021/ja067417a |
| [52] |
M. Terada, K. Sorimachi, J. Am. Chem. Soc. 129 (2007) 292-293. DOI:10.1021/ja0678166 |
| [53] |
S.L. You, Q. Cai, M. Zeng, Chem. Soc. Rev. 38 (2009) 2190-2201. DOI:10.1039/b817310a |
| [54] |
F. Xu, D. Huang, C. Han, et al., J. Org. Chem. 75 (2010) 8677-8680. DOI:10.1021/jo101640z |
| [55] |
M.H. Zhuo, Y.J. Jiang, Y.S. Fan, et al., Org. Lett. 16 (2014) 1096-1099. DOI:10.1021/ol403680c |
| [56] |
A. Fu, W. Meng, H. Li, J. Nie, J.A. Ma, Org. Biomol. Chem. 12 (2014) 1908-1918. DOI:10.1039/c3ob42157k |
| [57] |
M.J. Ajitha, K.W. Huang, Org. Biomol. Chem. 13 (2015) 10981-10985. DOI:10.1039/C5OB01473E |
| [58] |
J. Pous, T. Courant, G. Bernadat, et al., J. Am. Chem. Soc. 137 (2015) 11950-11953. DOI:10.1021/jacs.5b08515 |
| [59] |
S. Romanini, E. Galletti, L. Caruana, et al., Chem.-Eur. J. 21 (2015) 17578-17582. DOI:10.1002/chem.201502655 |
| [60] |
P.A. Champagne, K.N. Houk, J. Am. Chem. Soc. 138 (2016) 12356-12359. DOI:10.1021/jacs.6b08276 |
| [61] |
Y.Y. Khomutnyk, A.J. Arguelles, G.A. Winschel, et al., J. Am. Chem. Soc. 138 (2016) 444-456. DOI:10.1021/jacs.5b12528 |
| [62] |
T.J. Seguin, S.E. Wheeler, ACS Catal. 6 (2016) 7222-7228. DOI:10.1021/acscatal.6b01915 |
| [63] |
T. Wang, Z. Yu, D.L. Hoon, et al., J. Am. Chem. Soc. 138 (2016) 265-271. DOI:10.1021/jacs.5b10524 |
| [64] |
L. Simon, J.M. Goodman, J. Am. Chem. Soc. 131 (2009) 4070-4077. DOI:10.1021/ja808715j |
| [65] |
L. Simon, J.M. Goodman, J. Org. Chem. 75 (2010) 589-597. DOI:10.1021/jo902120s |
| [66] |
C. Zheng, Y.F. Sheng, Y.X. Li, S.L. You, Tetrahedron 66 (2010) 2875-2880. DOI:10.1016/j.tet.2010.02.031 |
| [67] |
L. Simon, J.M. Goodman, J. Org. Chem. 76 (2011) 1775-1788. DOI:10.1021/jo102410r |
| [68] |
L.M. Overvoorde, M.N. Grayson, Y. Luo, J.M. Goodman, J. Org. Chem. 80 (2015) 2634-2640. DOI:10.1021/jo5028134 |
| [69] |
J.T.M. Correia, B. List, F. Coelho, Angew. Chem. Int. Ed. 56 (2017) 7967-7970. DOI:10.1002/anie.201700513 |
| [70] |
M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian 09, Revision D. 01, Gaussian, Inc, Wallingford, CT, 2013.
|
| [71] |
C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785-789. DOI:10.1103/PhysRevB.37.785 |
| [72] |
A.D. Becke, J. Chem. Phys. 98 (1993) 5648-5652. DOI:10.1063/1.464913 |
| [73] |
A.V. Marenich, C.J. Cramer, D.G. Truhlar, J. Phys. Chem. B 113 (2009) 6378-6396. DOI:10.1021/jp810292n |
| [74] |
X. Qi, S. Liu, T. Zhang, et al., J. Org. Chem. 81 (2016) 8306-8311. DOI:10.1021/acs.joc.6b01429 |
| [75] |
C. Shan, X. Luo, X. Qi, et al., Organometallics 35 (2016) 1440-1445. DOI:10.1021/acs.organomet.6b00064 |
| [76] |
L. Zhu, X. Qi, Y. Lan, Organometallics 35 (2016) 771-777. DOI:10.1021/acs.organomet.6b00007 |
| [77] |
Y. Li, L. Zou, R. Bai, Y. Lan, Org. Chem. Front. 5 (2018) 615-622. DOI:10.1039/C7QO00850C |
| [78] |
X. Yue, C. Shan, X. Qi, et al., Dalton Trans. 47 (2018) 1819-1826. DOI:10.1039/C7DT04084A |
| [79] |
Y. Zhao, D.G. Truhlar, Theor. Chem. Acc. 120 (2008) 215-241. DOI:10.1007/s00214-007-0310-x |
| [80] |
S. Liu, Y. Lei, X. Qi, Y. Lan, J. Phys. Chem. A 118 (2014) 2638-2645. |
| [81] |
X. Qi, H. Zhang, A. Shao, et al., ACS Catal. 5 (2015) 6640-6647. DOI:10.1021/acscatal.5b02009 |
| [82] |
S. Yu, S. Liu, Y. Lan, B. Wan, X. Li, J. Am. Chem. Soc. 137 (2015) 1623-1631. DOI:10.1021/ja511796h |
| [83] |
X. Qi, Y. Li, R. Bai, Y. Lan, Acc. Chem. Res. 50 (2017) 2799-2808. DOI:10.1021/acs.accounts.7b00400 |
| [84] |
E.R. Johnson, S. Keinan, Mori-Sanchez P., et al., J. Am.Chem. Soc. 132 (2010) 6498-6506. DOI:10.1021/ja100936w |
| [85] |
Contreras-Garcia J., E.R. Johnson, S. Keinan, et al., J. Chem. Theory Comput. 7 (2011) 625-632. DOI:10.1021/ct100641a |
| [86] |
T. Lu, F. Chen, J. Comput. Chem. 33 (2012) 580-592. DOI:10.1002/jcc.v33.5 |
| [87] |
C.X. Cui, Z.P. Zhang, L. Zhu, et al., Phys. Chem. Chem. Phys. 19 (2017) 30393-30401. DOI:10.1039/C7CP06365B |