 2018, Vol. 29
2018, Vol. 29 



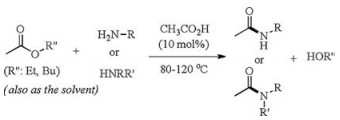
Due to the ready generation of ammonium carboxylate salts, amide bond formation by direct condensation of carboxylic acids and amines is challenging and often needs high temperatures or the presence of stoichiometric amounts of sacrificing coupling reagents [1, 2]. To solve this problem, great efforts have been made to construct amide bond from coupling of activated acyl and/or amino reagents [3]. Among the diverse strategies, aminolysis of active esters by amines is one attractive practical method, which is apparently a nucleophilic carbonyl substitution reaction with the replacement of an alkoxide by an amino group [4].
Very recently, Sharley and Williams reported an acetic acidcatalyzed aminolysis reaction of simple unactivated esters by amines to produce amides (Scheme 1) [5]. The most impressive achievement of this study is the use of simple acetic acid as the efficient catalyst to enable amide bond formation from common carboxylic esters. A range of amines, primary or secondary, can be coupled with acetate esters (as well as several other acid esters including formate, benzoate and amino acid ester) to give the amide products. The acetyl group in the amide products is found to originate from the ester, rather than from the acetic acid catalyst, based on the findings of using other acid acids as catalyst, stoichiometric amounts of acetic acid and 13C labeling experiments. As a result, a stepwise mechanism of nucleophilic addition of amine to carbonyl/C—O bond cleavage is invoked for this reaction, featuring an intermediate tetrahedral adduct. This classic stepwise mechanism is widely invoked in organic chemistry textbooks for nucleophilic acyl substitution reactions, and has been supported by some recent mechanistic and computational studies [6].

|
Download:
|
| Scheme 1. Amide bond forming reaction from aminolysis of ester by amine catalyzed by acetic acid. | |
{kind=link}
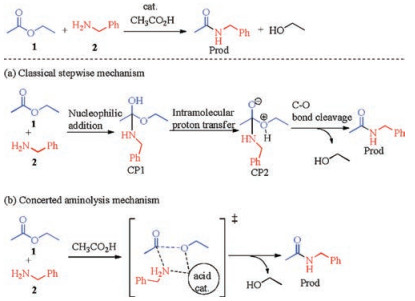
During our recent computational study on the mechanism of an ynamide-promoted amide bond formation [7], a carboxylic acidcatalyzed concerted aminolysis of active ester intermediate by amine is proposed as the key amide bond-forming step [8]. Under this mechanism, the formation of acyl-amino bond and the cleavage of acyl—O bond occur simultaneously, without the intermediacy of a tetrahedral adduct nor the rehybridization of the acyl carbon atom from sp2 to sp3. This mechanism is competitive with the classic stepwise mechanism involving the tetrahedral adduct intermediate. We therefore get interested into this acetic acid-catalyzed aminolysis reaction of alkyl ester with amines. In this manuscript, a detailed computational study is performed to elucidate the mechanism for this intriguing reaction. Particular focus is set on the establishment and comparison of the classic stepwise mechanism and the concerted aminolysis mechanism, and the specific role and reactivity pattern of acetic acid catalyst in this reaction.
All the calculations were performed using Gaussian 09 program [9]. All the stationary points were obtained using solution-phase geometry optimizations using B3LYP functional [10] in combination with a large triple-ζ 6-311G(d, p) basis set. To account for solvation effect, self-consistent reaction field (SCRF) theory [11] with CPCM solvation model [12] was used for geometry optimization in ethyl acetate (the solvent used in the experiments). For each optimized structure, frequency analysis was performed to confirm it is a minimum or transition state, and to get thermodynamic corrections. For each transition state, intrinsic reaction path analysis [13] was used to follow the reaction path in both directions and to obtain the reactant and product that this transition state connects. For each stationary point, its energy was calculated by solution-phase single point electronic energy plus free energy correction items. The energies of optimized intermediates and transition states are corrected to 1 mol/L at 298 K (a constant of 1.89 kcal/mol was added to the energy of each stationary point).
A real reaction of carboxylic ester 1 and amine 2 in the presence of acetic acid as catalyst was studied as the model reaction to elucidate the reaction pathways, as shown in Scheme 2. Two mechanisms have been studied in detail: (a) classic stepwise mechanism and (b) a concerted acyl substitution mechanism. The classic stepwise mechanism involves initial addition of amines to carbonyl of ester, followed by intramolecular proton transfer and C—O bond cleavage. In contrast, the concerted mechanism gives the amide product through a concerted process of replacement of alkoxide by an amino at the acyl group, which is promoted by bifunctional stabilizing interactions of the acid catalyst with both the amine and alkoxide moieties. The following sections show detailed results for the two mechanisms.

|
Download:
|
| Scheme 2. Model reaction and proposed mechanism profile. | |
{kind=link}
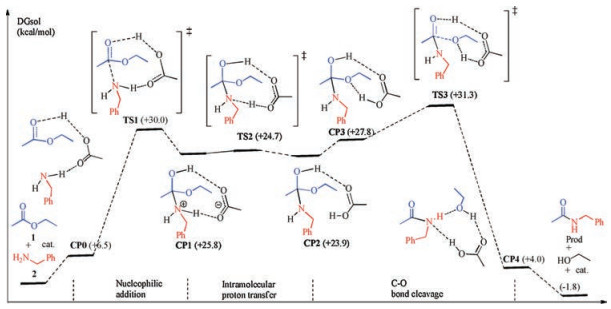
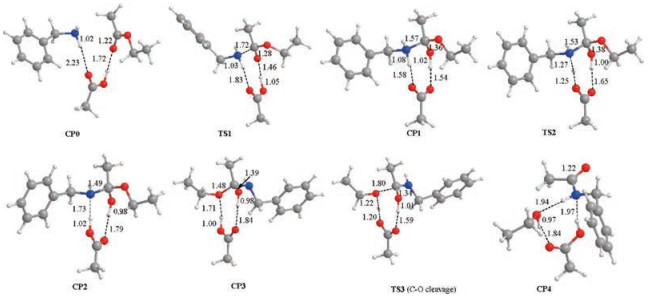
Scheme 3 and Fig. 1 show the reaction energy profile of a catalytic cycle for the classic stepwise mechanism and the key intermediates and transition states located (more details please see Supporting information). Calculation results show that nucleophilic addition of amine to ester has an activation free energy of 30.0 kcal/mol and is highly endergonic by 25.8 kcal/mol. A key transition state TS1 is located with acetic acid as the catalyst to activate the carbonyl group, which connects to a precomplex CP0 composed of ester, amine and the acetic acid catalyst, and a tetrahedral adduct CP1. CP1 is inherently an ammonium carboxylate ion-pair and the ester carbonyl atom rehybridizes from sp2 in CP0 to sp3 in CP1, as reflected by the significant elongation of C (carbonyl)—O distance of 1.22 Å, to 1.28 Å to 1.36 Å in CP0, TS1 and CP1 respectively.

|
Download:
|
| Scheme 3. Reaction energy profile of a catalytic cycle for stepwise mechanism. Values in parentheses are relative free energies in kcal/mol in ethyl acetate. | |
{kind=link}

|
Download:
|
| Fig. 1. Optimized key intermediates and transition states for stepwise mechanism. All the values marked are bond lengths in Å. C, H, O and N atoms are depicted in grey, white, red and blue respectively in the structures. | |
{kind=link}
Then, intramolecular proton transfer in CP1 through transition state TS2 delivers CP2 with the proton transfer from ammonium to the carboxylate, as reflected by the elongation of N-H distance from 1.08 Å to 1.27 Å to 1.73 Å, and the concurrent shortening of the O-H distance from 1.58 Å to 1.25 Å to 1.02 Å, going from CP1 through TS2 to CP2. Carboxylic acid forms two hydrogen bonds in CP2: O (carbonyl)…H…O and O…H…N (Fig. 1).
Finally, C—O(ethoxy) bond cleavage in CP2 produces a ternary complex CP4 composed of the desired amide Prod, ethanol and the acetic acid catalyst through transition state TS3. In TS3, C—O (ethoxy) bond is significantly elongated to 1.80 Å from 1.48 Å in precomplex CP3, and completely cleaved in CP4. Accompanying with the C—O bond cleavage is the rehybridization of the central carbon from sp3 back to sp2, as can be seen from the C—O distance of 1.39 Å to 1.34 Å to 1.22 Å in CP3, TS3 and CP4, respectively. More, there are two additional concurrent proton transfer processes during the C—O bond cleavage, namely, one proton transfers from carboxylic acid to ethoxy while another proton transfers from hydroxy to carboxylate. Therefore, the carboxylate acts also as a proton shuttle during the C—O bond cleavage process. These proton transfer processes are evident by the changes of the two pairs of O…H…O contacts in CP3, TS3 and CP4 (Fig. 1). In CP4, multiple hydrogen bonds are observed between the amide product, ethanol and the acetic acid catalyst. Dissociation of CP4 gives the separated products, regenerates the catalyst, and thus completes a full catalytic cycle. Overall, this stepwise mechanism has an apparent activation free energies of 31.3 kcal/mol while is only slightly exergonic by 1.8 kcal/mol, indicating a weak thermodynamic driving force for this reaction.
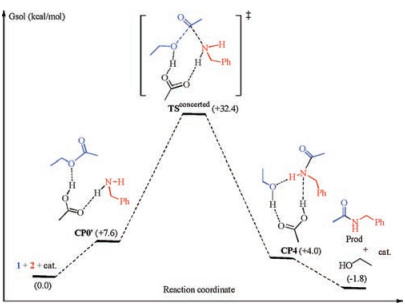
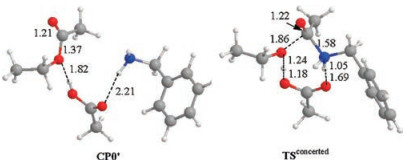
Scheme 4 and Fig. 2 show the results of the concerted acyl substitution mechanism. As can be seen, this mechanism features a key transition state TSconcerted in which the C(acyl)—O bond cleavage and C(acyl)—N bond formation occur concurrently, without the need of a tetrahedral adduct intermediate and the ensuing rehybridization of the acyl carbon atom. The keep of the sp2 hybridization state of the acyl carbon is reflected by the short C (acyl)—O bond of carbonyl and the double bond character. It is noteworthy that TSconcerted connects directly with the precomplex CP0' and CP4 as verified by IRC analysis, confirming the absence of a tetrahedral adduct intermediate along the reaction coordinate of this concerted mechanism. This mechanism has an overall activation free energy of 32.4 kcal/mol.

|
Download:
|
| Scheme 4. Concerted acyl substitution mechanism. Values in parentheses are relative free energies in kcal/mol in ethyl acetate. | |
{kind=link}

|
Download:
|
| Fig. 2. Optimized key intermediates and transition states for concerted aminolysis mechanism. All the values marked are bond lengths in Å. C, H, O and N atoms are depicted in grey, white, red and blue respectively in the structures. | |
{kind=link}
With the catalytic cycles for both mechanisms established, it may now be possible to make a comparison. Indeed, our calculation results show that these two mechanisms have very similar overall activation barriers (31.3 versus 32.4 kcal/mol), and therefore may probably compete with each other in the reaction course. However, considering the retention of stereo-chemistry of chiral centers of both amines and carboxylic esters moieties in the amide products, it is strongly proposed that the concerted aminolysis mechanism is more likely to be the operating mechanism because this mechanism is a one-step concerted process which does not need the rehybridization of the central acyl carbon. In contrast, the classic stepwise mechanism occurs with rehybridization of the acyl carbon from sp2 to sp3 and back to sp2 again. The rehybridization of the carbonyl carbon can generate/ eliminate new chiral centers that may exert significant influence on the neighboring existing stereocenters in asymmetric esters and/or amines, and may thus lead to epimerization/racemization of the final amide products [14]. Therefore, the reaction studied is proposed to be more prone to follow the concerted aminolysis mechanism without the intermediacy of a tetrahedral adduct, nor the rehybridization of the acyl carbon. Nevertheless, the classical stepwise mechanism cannot be completely ruled out.
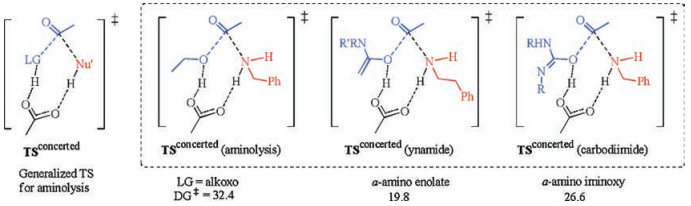
Moreover, from these results as well as those mechanistic proposals for related amide forming reactions in the presence of different coupling reagents, some inherent relationship may now be evident. Reactions using classic carbodiimide and the recent ynamide coupling reagent may all be accommodated into this concerted aminolysis mechanism where the active ester intermediates have different leaving group OR with R being alkyl, α-amino vinyl or α-amino iminyl (Fig. 3). For example, carbodiimide would have an α-amino iminyl group on the oxygen, namely an α-amino iminyl ester is involved as the active ester that undergoes the concerted aminolysis by amine. Calculation results of dicyclohexyl carbodiimide has an overall activation free energy of 26.6 kcal/mol for the aminolysis of the active ester by benzyl amine, which is kinetically feasible and in part supports the possible involvement of concerted acyl substitution mechanism for reactions using carbodiimide as the coupling reagent (please see Supporting information for more details). For another example, the ynamide coupling reagent recently developed is also suggested to follow direct concerted aminolysis of active α-amino vinyl ester by amine, with a low activation free energy of only 19.8 kcal/mol [7, 8]. These results show that the p-π conjugation between the R group with the oxygen atom in leaving group OR may be a very important factor for stabilizing the transition state of aminolysis. These results and implications may open up new opportunities for the tuning and development of active coupling reagent for amide bond formation as well as other related acyl substitution reactions.

|
Download:
|
| Fig. 3. A general concerted acyl substitution mechanism scaffold proposed. Values are activation free energies in kcal/mol. | |
{kind=link}
In summary, the reaction mechanism for acetic acid-catalyzed ester aminolysis is studied by computational evaluation of the classic stepwise mechanism and a concerted acyl substitution mechanism. The concerted acyl substitution mechanism is proposed to be probable that may be widely operating in a variety of related amidation reactions with different types of coupling reagents.
AcknowledgmentsThis study was supported by the National Natural Science Foundation of China (No. 21472068). Financial support from MOE & SAFEA for the 111 Project ([No. B13025) is also gratefully acknowledged.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.01.003.
| [1] |
(a) E. Valeur, M. Bradley, Chem. Soc. Rev. 38 (2009) 606-631 (b) C. L. Allen, J. M. J. Williams, Chem. Soc. Rev. 40 (2011) 3405 (c) V. R. Pattabiraman, J. W. Bode, Nature 480 (2011) 471-479 (d) H. Lundberg, F. Tinnis, N. Selander, H. Adolfsson, Chem. Soc. Rev. 43 (2014) 2714-2742. |
| [2] |
(a) A. El-Faham, F. Albericio, Chem. Rev. 111 (2011) 6557-6602 (b) J. C. Sheehan, G. P. Hess, J. Am. Chem. Soc. 77 (1955) 1067-1068 (c) G. Gawne, G. W. Kenner, R. C. Sheppard, J. Am. Chem. Soc. 91 (1969) 5669-5671 (d) L. A. Carpino, P. Henklein, B. M. Foxman, et al., J. Org. Chem. 66 (2001) 5245-5247. |
| [3] |
(a) J. W. Bode, R. M. Fox, K. D. Baucom, Angew. Chem. Int. Ed. 45 (2006) 1248-1252 (b) B. Shen, D. M. Makley, J. N. Johnston, Nature 465 (2010) 1027-1032 (c) W. Wu, Z. Zhang, L. S. Liebeskind, J. Am. Chem. Soc. 133 (2011) 14256-14259 (d) G. M. Fang, Y. M. Li, F. Shen, et al., Angew. Chem. Int. Ed. 50 (2011) 7645-7649 (e) J. F. Soulé, H. Miyamura, S. Kobayashi, J. Am. Chem. Soc. 133 (2011) 18550-18553 (f) A. M. Dumas, G. A. Molander, J. W. Bode, Angew. Chem. Int. Ed. 51 (2012) 5683-5686 (g) J. X. Wang, G. M. Fang, Y. He, et al., Angew. Chem. Int. Ed. 54 (2015) 2194-2198 (h) J. Li, M. J. Lear, Y. Kawamoto, et al., Angew. Chem. Int. Ed. 54 (2015) 12986-12990 (i) H. Noda, J. W. Bode, J. Am. Chem. Soc. 137 (2015) 3958-3966. |
| [4] |
(a) C. G. McPherson, N. Caldwell, C. Jamieson, I. Simpson, A. J. B. Watson, Org. Biomol. Chem. 15 (2017) 3507-3518 (b) C. Sabot, K. A. Kumar, S. Meunier, C. Mioskowski, Tetrahedron Lett. 48 (2007) 3863-3866 (c) N. Caldwell, C. Jamieson, I. Simpson, A. J. B. Watson, Chem. Commun. 51 (2015) 9495-9498. |
| [5] |
D.D.S. Sharley, J.M.J. Williams, Chem. Commun. 53 (2017) 2020-2023. DOI:10.1039/C6CC09023K |
| [6] |
H. Charville, D.A. Jackson, G. Hodges, A. Whiting, M.R. Wilson, Eur. J. Org. Chem. (2011) 5981-5990. |
| [7] |
(a) T. Krause, S. Baader, B. Erb, L. J. Gooßen, Nat. Commun. 7 (2016) 11732 (b) L. Hu, S. Xu, Z. Zhao, et al., J. Am. Chem. Soc. 138 (2016) 13135-13138. |
| [8] |
S.L. Zhang, H.X. Wan, Z.Q. Deng, Org. Biomol. Chem. 15 (2017) 6367-6374. DOI:10.1039/C7OB01378G |
| [9] |
M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian 09, Revision D. 01, Gaussian, Inc., Wallingford, CT, 2013.
|
| [10] |
(a) A. D. Becke, J. Chem. Phys. 98 (1993) 5648-5652 (b) C. Lee, W. Yang, R. G. Parr, Phys. Rev. B 37 (1988) 785-789. |
| [11] |
M. Cossi, N. Rega, G. Scalmani, V. Barone, J. Compt. Chem. 24 (2003) 669-681. DOI:10.1002/jcc.10189 |
| [12] |
(a) J. Tomasi, B. Mennucci, R. Cammi, Chem. Rev. 105 (2005) 2999-3093 (b) Y. Fu, L. Liu, R. Q. Li, R. Liu, Q. X. Guo, J. Am. Chem. Soc. 126 (2004) 814-822. |
| [13] |
(a) C. Gonzalez, H. B. Schlegel, J. Phys. Chem. 94 (1990) 5523-5527 (b) K. Fukui, Acc. Chem. Res. 14 (1981) 363-368. |
| [14] |
G.W. Anderson, F. Callahan, J. Am. Chem. Soc. 80 (1958) 2902-2903. DOI:10.1021/ja01544a077 |