 2018, Vol. 29
2018, Vol. 29 



b School of Chemical Engineering and Light Industry, Guangdong University of Technology, Higher Education Mega Center, Guangzhou 510006, China
Nowadays, energy shortage is considered to be one of the major challenges in sustainable development world. A top priority to resolve energy problems is searching for alternative renewable energy for traditional fossil energy. Due to the high-efficient, environmental-benign and abundant resources, hydrogen energy has attracted great attention [1-4]. However, the practical application of hydrogen is inhibited by the challenge in hydrogen storage. In general, there are two strategies for hydrogen storage, i.e., chemical and physical methods. Compared to the physical method, chemical hydrogen storage is more secure and efficient. Unfortunately, chemical hydrogen storage requires activation of the unreactive H—H bond, which is extremely difficult [5-7]. Therefore, developing high-efficient catalysts for hydrogenation activation is of critical importance and has become a hot topic in the field of hydrogen storage and usage.
Recently, scientists proposed a new type of bifunctional metalfree catalysts named frustrated Lewis pairs (FLPs) on the basis of classical Lewis' theory of acids and bases, providing a novel strategy for hydrogen storage [8-10]. FLPs are cooperative catalysts combined by Lewis acids (LA) and Lewis bases (LB) without selfquenching (Fig. 1a). Due to the electronic effect and steric hindrance, the LA and LB of FLPs remain independent rather than forming classical Lewis adduct. As a result, LA and LB are able to serve as electron accepter and donor simultaneously, which endows FLPs unprecedented bifunctional properties as transition metal for catalyzed activation of small molecules. In 2006, Stephan et al. reported a reversible H2 activation reaction using an FLP, (Mes)2P-C6F5-B(C6F5)2 (Mes = 2, 4, 6-trimethylphenyl) [8]. In the past decade, the applications of FLPs have extended to hydrogenation of unsaturated bonds [11-13], H—E bond activation [14-16], capture and reduction of small molecules [17-19], polymerization [20-22] and other scopes of chemistry. FLP has become one of the most successful metal-free catalytic systems.

|
Download:
|
| Fig. 1. (a) Structure of frustrated Lewis pairs; (b) H2 cleavage by frustrated Lewis pairs; (c) General view of kinetics and thermodynamics in FLPs catalyzed H2 activation. | |
In 2007, Pápai and coworkers performed a detailed atomistic insight into the mechanism of hydrogen activation and proposed a synergistic electron transfer (ET) model, which has been widely accepted [23]. As exhibited in Fig. 1b, when H2 is close to the cavity formed by LA and LB, the σ(H—H) orbital would interact with the empty orbital of LA while LB donates the lone pair to the σ*(H—H) orbital, leading to weakening and, ultimately, to the heterolytic cleavage of H—H bond. The energy barrier of this model mainly originates from creating the orbital overlaps and distorting the individual LA and LB moieties.
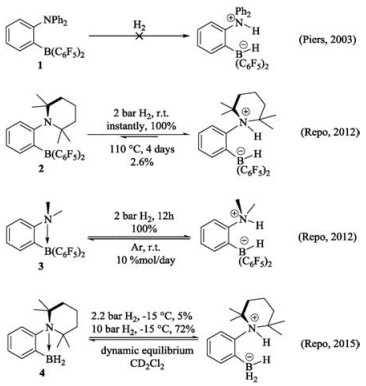
The bifunctional catalytic strategy for hydrogenation and dehydrogenation has attracted our continued interests recently [24-30]. With respect to catalyst design, reversible hydrogen activation is extremely important in terms of 1) the development of system capable of both hydrogen storage and release, 2) the development of catalysts for (de)hydrogenation. However, not all bifunctional FLPs could activate hydrogen reversibly, not only because of the free energy of activation, but also because the energy of dihydrogen adduct is often unsuitable (too high to unstable or too low to turn back, Fig. 1c). Herein, only some of FLPs could be used as catalysts for reversible H2 activation (Fig. 2). In 2003, Piers et al. synthesized an ansa-aminoborane ortho-NPh2- C6H4-B(C6F5)2 (1). The complex 1 could react with small acids (e.g., H2O and HCl) with the formation of zwitterionic products, however, it failed to successfully catalyze H2 activation, due to the low basicity of the nitrogen center with diphenylamino moiety [31]. Until 2012, Repo and co-workers reported other two aminoboranes, i.e., ortho-TMP-C6H4-B(C6F5)2 (TMP = 2, 2, 6, 6-tetramethylpiperidine) (2) and ortho-NMe2-C6H4-B(C6F5)2 (3), which have the same linker and LA moiety but different LB moiety as original aminoborane 1 [32]. Both 2 and 3 readily activate H2 at mild conditions (2 bar H2) and form corresponding ammonium borohydrides. However, only adduct of 3 could turn back to reactants, while product of 2 remains stable even is heated for 4 days. In 2015, Repo reported an analogue of 3, ortho-TMP-C6H4-BH2 (4), by replacing C6F5 with H [33]. It is also found that 4 could reversibly activate H2 under moderate pressures and temperatures, and the exchange rate is faster than that of 3.

|
Download:
|
| Fig. 2. Four types of reported FLPs with similar structures and their H2 activation reactions. | |
It is interesting that the reactivity of FLPs 1-4 for hydrogen activation is distinguishable, although the structures are similar. This dramatic reactivity difference inspires us to explore the intrinsic relationship between structure and the reactivity of FLPs, which is of crucial importance to design more efficient FLPs based metal-free catalysts for hydrogen activation and storage. In present report, a systemic DFT study (details in Supporting information) was carried out to understand the mechanism of these four types of FLPs catalyzed H2 activation. The intrinsic factors of the reactivity of FLPs catalyzed hydrogen activation was analyzed in terms of kinetics and thermodynamics. Moreover, a serial of FLPs with phenyl derivatives as LA groups was designed to reveal how the electronic properties of the substituents affect the reactivity of FLPs. Present systemic investigations should provide useful guidelines for design of efficient FLPs catalysts for reversible hydrogen activation.
Firstly, the detailed mechanism of FLPs 1-4 catalyzed H2 activation was explored according to the electron transfer (ET) model [10]. The free energy profiles of hydrogen activation catalyzed by FLPs 1 and 2 are depicted in Fig. 3a, while the free energy profiles for 3 and 4 are shown in Fig. 3b. Since it has suggested that the polarity of solvent has significant influence on the FLPs catalyzed H2 activation, the solvent effect of both CH2Cl2 and toluene are considered in the following discussions.

|
Download:
|
| Fig. 3. The free energy profiles of hydrogen activation catalyzed by FLP catalysts (a) 1 and 2; (b) 3 and 4. Relative Gibbs free energies (in kcal/mol) are correlated with solvation effect of CH2Cl2 (front) and toluene (back), respectively. | |
As shown in Fig. 3a, when ortho-NPh2-C6H4-B(C6F5)2 (1) is employed to catalyze H2 activation, it would form intermediate im-1 firstly, then the H-H cleavage occurs at a late transition state (H—H = 0.89 Å, N—H = 1.52 Å, B—H = 1.39 Å), as compared with other three FLPs. This could attribute to the weak basicity of the nitrogen center with phenyl groups, which is unfavorable for the interaction between LB and σ*(H-H) orbital in a remote distance. The calculated free energy of im-1 is 17.5 kcal/mol in CH2Cl2 (17.9 kcal/mol in toluene) relative to the separated reactants. Then with the assistance of the lone pair electrons of N atom, FLP catalyst prompts the cleavage of H—H bond. The corresponding transition state is ts-1 with the free energy barrier of 18.4 kcal/mol (18.6 kcal/mol in toluene), which is a late transition state and close to the product. Unfortunately, the product p-1 is endergonic by more than 6.8 kcal/mol and 8.5 kcal/mol in CH2Cl2 and toluene respectively, which indicates the H2 activation catalyzed by 1 is difficult to occur in terms of thermodynamics. The calculated results are in good agreement with the experimental observation that FLP 1 failed to successfully catalyze H2 activation [31].
In the case of ortho-TMP-C6H4-B(C6F5)2 (2), the reaction pathway is slightly different from catalysts 1. The empty orbital of LA center and the lone pair of LB center interact with the σ(H—H) bond and σ*(H-H) of H2 simultaneously, leading to a cooperative H2 cleavage transition state ts-2. The intermediate of H2-LA complex could not be located, probably attributed to the stronger LB center in FLP 2 compared with FLP 1. The free energy of ts-2 (18.1 kcal/mol and 17.8 kcal/mol in CH2Cl2 and in toluene respectively) is similar as that of ts-1. However, the thermodynamics of this reaction is quite different with that of FLP 1. The product borohydride p-2 is exergonic by as large as 17.1 kcal/mol in CH2Cl2 (14.2 kcal/mol in toluene), indicating it is highly stable. It is obvious that FLP 2 catalyzed H2 activation is irreversible. The thermodynamic difference could ascribe to the different strength of LB center in FLP 1 and 2. Our calculation results well explain the experimental facts that FLP 2 readily activated H2 at mild conditions without reverse reaction observed even after heating for 4 days [32].
The ansa-aminoborane 3 was proved to be able to activate hydrogen reversibly in Repo's experiments [32]. Interestingly, due to the small steric hindrance of methyl groups, FLP 3 usually forms a classical Lewis adduct with four-membered B-N ring, as shown in Fig. 3b (dat-3). Before H2 hydrogen activation, the Lewis adduct dat-3 must decompose to unquenched FLP 3 by breaking the B-N interaction. Fortunately, FLP 3 is only 3.7 and 3.2 kcal/mol less stable than Lewis adduct dat-3 in CH2Cl2 and toluene thermodynamically. FLP 3 then reacts with H2 via concerted transition state ts-3 to yield borohydride product p-3. Similar to catalyst 2, 3 cannot form a stable H2-LA complex either. The free energy barrier of ts-3 is 23.4 kcal/mol in CH2Cl2, and 24.8 kcal/mol in toluene, which is a little higher than those of catalysts 1 and 2. The borohydride product p-3 is slightly lower than dat-3 by -4.4 kcal/mol+ in CH2Cl2 (-1.7 kcal/mol in toluene), indicating the reaction is thermodynamically favorable. Moreover, the reversed dehydrogenation is also feasible as the free energy barrier is not very high (ΔG= 27.8/26.5 kcal/mol). Therefore, it is reasonable to conclude that FLP 3 could catalyzed hydrogen activation reversibly, which is in good agreement with the experimental results [32].
As for the catalyst ortho-TMP-C6H4-BH2 (4), it is the analogue of 2 bearing a smaller LA site with weaker Lewis acidity [33]. According to the experimental results, FLP 4 exists as a trans dimer di-4 in the solid state. And the dimer di-4 should be in equilibrium with the monomeric dat-4 with dative B-N interaction in solution. Intermediate dat-4 needs to further overcome the dative B-N interaction to form an open monomer 4 from dimer before reacting with H2. Then the H2 is cooperatively cleaved by 4 via a concerted transition state (ts-4) to form the borohydride product p-4. The free energy of ts-4 is 22.3 and 22.2 kcal/mol in CH2Cl2 and toluene, which is 1.1 kcal/mol and 2.6 kcal/mol lower than that of ts-3, respectively. It is intriguing that the thermodynamic of this reaction is of significant difference in CH2Cl2 and toluene. On the one hand, the borohydride product p-4 locates only 0.4 kcal/mol below reactants in CH2Cl2, which implies the hydrogenation activation and dehydrogenation is almost in equilibrium. In other words, FLP 4 is able to catalyze the hydrogen activation reversibly. On the other hand, the zwitterionic product is endergonic by 4.2 kcal/mol in low polar solvent, which is accordance with Repo's experimental results [33].
Usually, without intrinsic interaction between the LA and the LB sites, late TS would lead to an endergonic process for H2 activation. In contrast, the reaction is usually exergonic with early TS. However, the interaction of LA-LB (extra deformation energy) should be included when estimating the reversibility of the H2 activation. In order to understand the intrinsic influence of geometric and electronic effects on the reactivity of FLPs catalyzed H2 activation, and provide useful guidelines for improving the catalytic efficient of FLPs rationally, it is necessary to analyze the composition of the free energy.
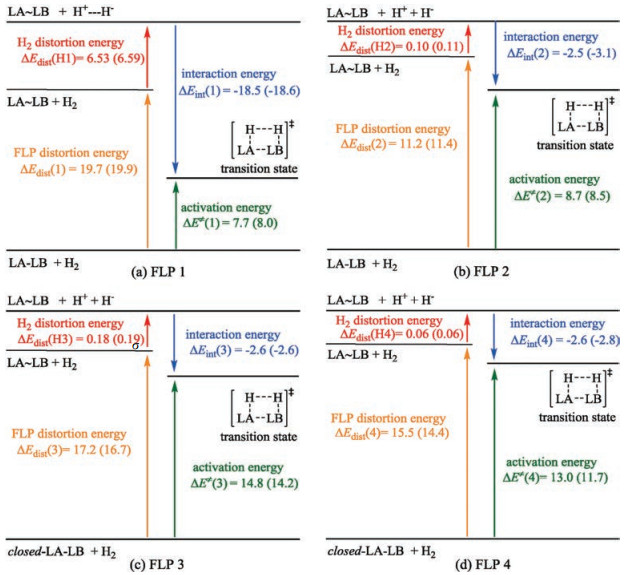
According to the framework of distortion/interaction model proposed by Morokuma [34], Houk [35] and Bickelhaupt [36, 37], the energies of transition states for FLPs catalyzed H2 activation were partitioned into distortion energy (Edis) and interaction energy (Eint) as shown in Fig. 4. Therefore, the activation energy could be defined as E = Edis + Eint. The distortion energy of FLP 1, ΔEdis(1) and Edis(H1) are much higher than the others due to asymmetric LB group (Fig. S1 in Supporting information) and late transition state. However, the interaction energy of FLP 1 is the largest (-18.5/-18.6 kcal/mol) attributed to the unstable fragments of severe distorted reactants. As a result, the activation energy of FLP 1 catalyzed reaction is only 7.7 and 8.0 kcal/mol in CH2Cl2 and toluene. The distortion energies of FLP 3 and 4 also include extra energy used to break quenched N—B bond, which increases the energy of decomposition and activation. It is obvious that ΔEHH of FLP 2, 3 and 4 is much small, which indicates H2 molecular has strong interaction and processes an early transition state. However, ΔEHH of FLP 1 is high because of the low basicity of LB. As results, based on interaction energy of FLP 2-4, there is no obvious difference of kinetic energy of four FLPs, though they have significant diverse LA/LB substituents with different electronic effect.

|
Download:
|
| Fig. 4. Kinetic energy decomposition of H2 activation catalyzed by four FLPs. Zero-point energies (in kcal/mol) are correlated with solvation effect of CH2Cl2 (outside brackets) and toluene (inside brackets), respectively. | |
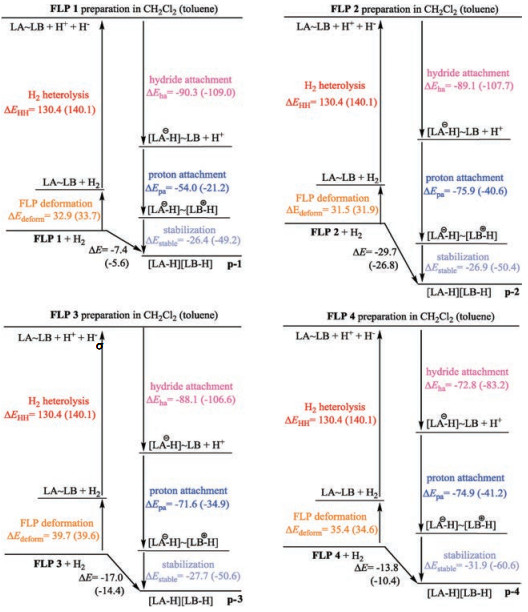
The overall thermodynamic energy of catalyzed H2 activation was divided into FLP preparation (ΔEdeform), H2 heterolytic cleavage (ΔEHH), hydride attachment (ΔEha), proton attachment (ΔEpa) and stabilization energies (ΔEstable) according to previous research [38]. For quenched FLP, ΔEdeform also contains the energy for N—B bond deformation. The overall thermodynamic energies then could be defined as the total of these five parts. As can be seen in Fig. 5, the ΔEHH for all FLPs is identical and the difference of ΔEstable is marginal. In contrast, the ΔEpa and ΔEha are distinguishable, indicating ΔEpa and ΔEha is of critical influence on the reactivity of FLPs. With the strong electron-withdrawing group -C6F5 as LA substituents, FLP 1, 2 and 3 have a high ability to accept the electron of σ(H—H) bond, thus the interaction between these FLPs and H2 are stronger than FLP 4 with -BH2 group as LA. The strong interaction ultimately leads to the high ΔEha. On the other hand, with electron-withdrawing group phenyls acting as LB, the strength of Lewis basicity of N atom in FLP 1 will reduce, which makes the interaction of σ*(H-H) orbital with the electron of LB weakened. As a result, the ΔEpa of FLP 1 is smaller than that of FLP 2, 3 and 4 with electron-donating group in LB moiety. As a result, the overall thermodynamic energy of FLP 1 catalyzed H2 activation is the smallest. It is in good accordance with the experimental results, which proved that FLP 1 is inert in the H2 activation [31].

|
Download:
|
| Fig. 5. Thermodynamic energy decomposition of H2 activation catalyzed by four FLPs. Zero-point energies (in kcal/mol) are correlated with solvation effect of CH2Cl2 (outside brackets) and toluene (inside brackets), respectively. | |
It is worth noting that solvent effect has a significant influence on the catalyzed H2 activation as well. As shown in Fig. 5, the ΔEHH in CH2Cl2 is 9.7 kcal/mol smaller than that in toluene, indicating the polar solvent (CH2Cl2) is more beneficial for the formation of zwitterionic molecule. Moreover, although the overall thermodynamic energy is different by less than 3 kcal/mol, the value of ΔEha, ΔEpa and ΔEstable in CH2Cl2 are different dramatically. Besides, because the dihydrogen product is in its zwitterionic form, these products are more stable in CH2Cl2 than in toluene. On the other hand, reactants are in their neutral forms, thus the polarity of solvent has minor effects on the reactants. As a result, the solvent effect on products is more dramatic than reactants to some extent (Table S3 in Supporting information). As for the transition states, the solvent effect is negligible as they are neutral as the reactants (Fig. 4). Combined the discussions above, it is reasonable that the effect of solvent is crucial for thermodynamics rather than kinetics.
After understanding of above results, further design of FLP catalysts through substituent effect is needed. In general, the reactivity of FLPs on H2 activation is mainly influenced by the property of LA and LB. The former accepts the electron of the σ(H-H) by the empty orbital, while the latter donates the lone pair to σ*(H-H) orbital. It should be pointed out that previous studies on the reactivity and design of FLPs mainly focus on the modifying the structure of LA [39], as the influence of LA is prominent than that of LB. Hence, we are going to evaluate and design FLPs through modifying the structure of LA in the following section, aiming to improve the performance of FLPs in catalyzed reversible H2 activation under mild conditions.
It is worth noting that the steric hindrance of LA and LB groups would affect the kinetics and thermodynamics of FLPs catalyzed H2 activation as well. For example, with reduced size of -NMe2 and -BH2 group, catalysts 3 and 4 usually appear in the form of stable traditional Lewis adducts. Therefore, additional energy is required to overcome in order to form a genuine type of FLP. In contrast, FLP 1 and 2 with bulky groups could constrain the LA-LB dative interaction or borane dimerization, resulting in a low free energy barrier for H2 activation (about 3–7 kcal/mol lower). The extra energy also effect equilibrium between the reactants and the zwitterionic adduct. In a word, the steric hindrance is a very important factor in designing high-efficient FLPs for catalyzed H2 activation.
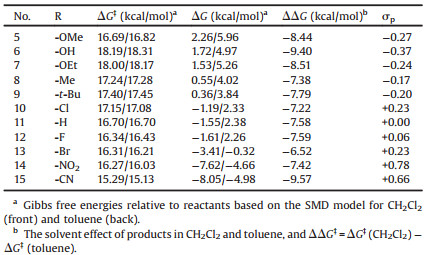
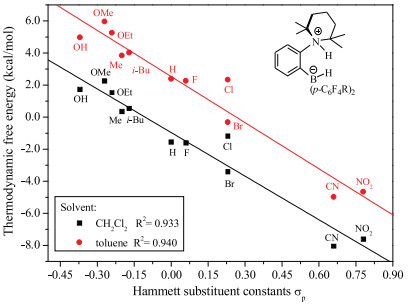
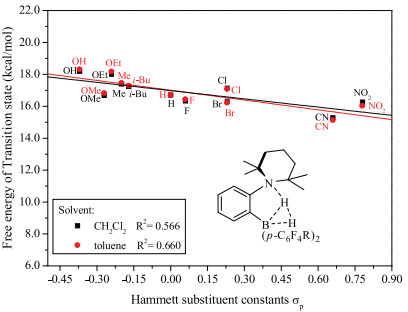
Based on above discussion, we utilized TMP (2, 2, 6, 6-tetramethylpiperidine) with good symmetry and basicity as LB substituent group, which is eligible electron-donating group and would avoid self-quenching. The LA and LB groups are combined by the short ortho-phenylene linker, so the basic skeleton is ortho-TMP-C6H4-B(p-C6H4R)2. Due to the easy modification of the electronic effect on phenyl group by adjusting its various substituted groups, a series of typical para-substituted phenyl derivatives are used as LA substituents. The free energy barrier (ΔG) and thermodynamic energy (ΔG) of these designed FLPs catalyzed H2 activation were calculated according the mechanism discussion above (Table 1). It is well-known that the electronic effects of the para-substituents on the reaction rate could be represented by Hammett substituent constant (σp) [40, 41], therefore, the relationship between ΔG and σp as well as that of ΔG and σp were evaluated and the results were depicted in Figs. 6 and 7, respectively.
|
|
Table 1 Kinetics (ΔG) and thermodynamics (ΔG) of FLPs catalyzed H2 activation and Hammett substituent constants. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
As shown in Fig. 6, there was a good linear relationship between ΔG and σp. The thermodynamic energy of H2 activation decreases with the σp of substituent increasing. The more exergonic of the reaction, the more difficult for the reaction to take place reversibly. For example, in the case of—CN (entry 15, Table 1), the thermodynamic energy is as large as -8.05 kcal/mol in CH2Cl2, thus the borohydride product is very stable, which hinders the reverse reaction to occur. The results indicate that the electronic effect of LA substituent has significant influence on the thermodynamics of FLP catalyzed H2 activation. On the other hand, though there is a poor linear relationship between free energy barrier (ΔG) and σp of substituent as shown in Fig. 7, the ΔG changes marginally with the sp changing from -0.37 to 0.78. It is obvious that electronic effects of LA substituent have negligible influence on the free energy barrier of FLP catalyzed H2 activation. These results are extremely interesting as we could achieve the reversible H2 activation through modifying the electronic property of LA.

|
Download:
|
| Fig. 6. The relationship between Hammett substituent constants and thermodynamic energy of dihydrogen adducts in two different solvents. | |
{kind=link}

|
Download:
|
| Fig. 7. The relationship between Hammett substituent constants and free energy barrier of FLPs catalyzed H2 activation in two different solvents. | |
{kind=link}
In conclusion, through controlling the energy of hydride attachment by adjusting substituents of LA, we could govern the thermodynamics of reversible H2 activation. Particularly, when the thermodynamic energy of reaction is zero, the reactions of H2 activation and dehydrogenation will become very easy to control in mild conditions. What is more, this adjustment has little effect on reaction rate, since it does not change the energy barrier fiercely (less than 2 kcal/mol).
In summary, a DFT study was performed to focus on the reaction path and thermodynamics of reversible H2 activation catalyzed by four previous reported FLPs catalysts. By comparison of four reported FLP structures and H2 activation, it could be concluded that the cooperation of LA and LB moieties plays an important role in the reversible H2 activation. On the one hand, LB group mainly contributes to the proton attachment, and it probably decides whether FLP could efficiently activate H2. On the other hand, LA group mainly controls the process of hydride attachment and it has a more significant effect on the thermodynamics of H2 activation, which mainly influences the reverse H2 liberation. The dimer and quenched structure of FLP also have a degree of influence on the performance of catalyzed H2 activation. Besides, polar solvent is beneficial for stabilizing reactants and zwitterionic dihydrogen adducts, and this solvent effect is stronger for products.
FLPs catalyzed reversible H2 activation is crucial for controllable H2 storage and liberation, or further hydrogenation/dehydrogenation. Due to the easy modification of the electronic effect of the borane groups, a series of FLPs were designed with various para-substituted phenyl as the LA substituents, to evaluate their performances for reversible H2 activation. As a result, we found the thermodynamic energy of adducts have a good linear relationship with Hammett substituent constants. And the electronic effect of LA site has obvious impact on thermodynamics but insignificant effect on kinetics. It reveals it is a promising way to control the thermodynamics of FLPs catalyzed reversible H2 activation by adjusting substituents of LA, and to control the direction of this reversible reaction, without obvious change in the reaction rate. These results possibly provide an in-depth understanding of structural and electronic effects of FLP on the kinetics and thermodynamics of reversible H2 activation. Hammett substituent constants also provide a convenient way to estimate the performance of different FLPs, which could serve as good guidance to design potential FLPs catalysts for H2 storage/ liberation or for further hydrogenation/dehydrogenation
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21502023 and 21473261), the Guangdong Natural Science Funds for Distinguished Young Scholar (No. 2015A030306027), and the Guangdong Special Support Plan for High-Level Talent (No. 20153100042090537). Computing facilities were supported in part by the high performance grid computing platform of Sun Yat-sen University, the Guangdong Province Key Laboratory of Computational Science, and Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (No. U1501501).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.02.007.
| [1] |
S. Satyapal, J. Petrovic, C. Read, G. Thomas, G. Ordaz, Catal. Today 120 (2007) 246-256. DOI:10.1016/j.cattod.2006.09.022 |
| [2] |
L. Schlapbach, A. Zuttel, Nature 414 (2001) 353-358. DOI:10.1038/35104634 |
| [3] |
D. Xu, C. Shan, Y. Li, et al., Inorg. Chem. Front. 4 (2017) 1813-1820. DOI:10.1039/C7QI00459A |
| [4] |
D. Xu, C. Shan, R. Bai, Y. Lan, Chin. J. Org. Chem. 37 (2017) 1231-1236. DOI:10.6023/cjoc201701033 |
| [5] |
T. He, N.P. Tsvetkov, J.G. Andino, et al., J. Am. Chem. Soc. 132 (2010) 910-911. DOI:10.1021/ja908674x |
| [6] |
T. Kimura, N. Koiso, K. Ishiwata, S. Kuwata, T. Ikariya, J. Am. Chem. Soc. 133 (2011) 8880-8883. DOI:10.1021/ja203538b |
| [7] |
D.M. Camaioni, Ginovska-Pangovska B., G.K. Schenter, S.M. Kathmann, T. Autrey, J. Phys. Chem. A 116 (2012) 7228-7237. DOI:10.1021/jp3039829 |
| [8] |
G.C. Welch, R.R.S. Juan, J.D. Masuda, D.W. Stephan, Science 314 (2006) 1124. DOI:10.1126/science.1134230 |
| [9] |
D.W. Stephan, Org. Biomol. Chem. 6 (2008) 1535-1539. DOI:10.1039/b802575b |
| [10] |
D.W. Stephan, G. Erker, Angew. Chem. Int. Ed. 49 (2010) 46-76. DOI:10.1002/anie.200903708 |
| [11] |
D.W. Stephan, Org. Biomol. Chem. 10 (2012) 5740-5746. DOI:10.1039/c2ob25339a |
| [12] |
D. W. Stephan, G. Erker, Frustrated Lewis pair mediated hydrogenations, in: G. Erker, D. W. Stephan (Eds. ), Frustrated Lewis Pairs I: Uncovering and Understanding, Springer, Berlin, Heidelberg, 2013, pp. 85-110.
|
| [13] |
Y. Wang, W. Chen, Z. Lu, Z.H. Li, H. Wang, Angew. Chem. Int. Ed. 52 (2013) 7496-7499. DOI:10.1002/anie.201303500 |
| [14] |
M.A. Dureen, A. Lough, T.M. Gilbert, D.W. Stephan, Chem. Commun. (2008) 4303-4305. |
| [15] |
M.H. Holthausen, J.M. Bayne, I. Mallov, R. Dobrovetsky, D.W. Stephan, J. Am. Chem. Soc. 137 (2015) 7298-7301. DOI:10.1021/jacs.5b04109 |
| [16] |
M.A. Légaré, M.A. Courtemanche, Rochette É., F.G. Fontaine, Science 349 (2015) 513. DOI:10.1126/science.aab3591 |
| [17] |
A.E. Ashley, A.L. Thompson, O'Hare D., Angew. Chem. Int. Ed. 48 (2009) 9839-9843. DOI:10.1002/anie.v48:52 |
| [18] |
A.J.P. Cardenas, B.J. Culotta, T.H. Warren, et al., Angew. Chem. Int. Ed. 50 (2011) 7567-7571. DOI:10.1002/anie.201101622 |
| [19] |
R.C. Neu, E. Otten, A. Lough, D.W. Stephan, Chem. Sci. 2 (2011) 170-176. DOI:10.1039/C0SC00398K |
| [20] |
Y. Zhang, G.M. Miyake, E.Y.X. Chen, Angew. Chem. Int. Ed. 49 (2010) 10158-10162. DOI:10.1002/anie.201005534 |
| [21] |
Y. Zhang, G.M. Miyake, M.G. John, et al., Dalton Trans. 41 (2012) 9119-9134. DOI:10.1039/c2dt30427a |
| [22] |
Piedra-Arroni E., C. Ladavière, A. Amgoune, D. Bourissou, J. Am. Chem. Soc. 135 (2013) 13306-13309. DOI:10.1021/ja4069968 |
| [23] |
T.A. Rokob, A. Hamza, A. Stirling, T. Soós, I. Pápai, Angew. Chem. Int. Ed. 47 (2008) 2435-2438. DOI:10.1002/(ISSN)1521-3773 |
| [24] |
C. Hou, J. Jiang, S. Zhang, et al., ACS Catal. 4 (2014) 2990-2997. DOI:10.1021/cs500688q |
| [25] |
C. Hou, Z. Zhang, C. Zhao, Z. Ke, Inorg. Chem. 55 (2016) 6539-6551. DOI:10.1021/acs.inorgchem.6b00723 |
| [26] |
C. Hou, J. Jiang, Y. Li, C. Zhao, Z. Ke, ACS Catal. 7 (2017) 786-795. DOI:10.1021/acscatal.6b02505 |
| [27] |
Z. Zhang, Y. Li, C. Hou, C. Zhao, Z. Ke, Catal. Sci. Technol. 8 (2018) 656-666. DOI:10.1039/C7CY02012K |
| [28] |
Y. Li, C. Hou, J. Jiang, et al., ACS Catal. 6 (2016) 1655-1662. DOI:10.1021/acscatal.5b02395 |
| [29] |
Y. Li, J. Zhang, S. Shu, et al., Chin. J. Org. Chem. 37 (2017) 2187-2202. DOI:10.6023/cjoc201703002 |
| [30] |
J. Zhang, J. Lin, Y. Li, et al., Catal. Sci. Technol. 7 (2017) 4866-4878. DOI:10.1039/C7CY01217A |
| [31] |
R. Roesler, W.E. Piers, M. Parvez, J. Organomet. Chem. 680 (2003) 218-222. DOI:10.1016/S0022-328X(03)00384-X |
| [32] |
K. Chernichenko, M. Nieger, M. Leskela, T. Repo, Dalton Trans. 41 (2012) 9029-9032. DOI:10.1039/c2dt30926b |
| [33] |
K. Chernichenko, B. Kótai, I. Pápai, et al., Angew. Chem. Int. Ed. 54 (2015) 1749-1753. DOI:10.1002/anie.201410141 |
| [34] |
K. Kitaura, K. Morokuma, Int. J. Quantum Chem. 10 (1976) 325-340. DOI:10.1002/(ISSN)1097-461X |
| [35] |
K.N. Houk, R.W. Gandour, R.W. Strozier, N.G. Rondan, L.A. Paquette, J. Am. Chem. Soc. 101 (1979) 6797-6802. DOI:10.1021/ja00517a001 |
| [36] |
F.M. Bickelhaupt, J. Comput. Chem. 20 (1999) 114-128. DOI:10.1002/(ISSN)1096-987X |
| [37] |
F.M. Bickelhaupt, K.N. Houk, Angew. Chem. Int. Ed. 56 (2017) 10070-10086. DOI:10.1002/anie.201701486 |
| [38] |
T.A. Rokob, A. Hamza, I. Pápai, J. Am. Chem. Soc. 131 (2009) 10701-10710. DOI:10.1021/ja903878z |
| [39] |
D.J. Scott, M.J. Fuchter, A.E. Ashley, Chem. Soc. Rev. 46 (2017) 5689-5700. DOI:10.1039/C7CS00154A |
| [40] |
C. Hansch, A. Leo, R.W. Taft, Chem. Rev. 91 (1991) 165-195. DOI:10.1021/cr00002a004 |
| [41] |
D. Xu, X. Qi, M. Duan, et al., Org. Chem. Front. 4 (2017) 943-950. DOI:10.1039/C6QO00841K |