 2018, Vol. 29
2018, Vol. 29 



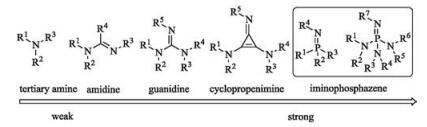
In the field of asymmetric catalysis, organocatalysis together with metal catalysis and enzymatic transformations are the three general categories to the catalytic production of stereochemical diversity. Among the different types of organocatalysts, chiral organobases [1-3], mainly nitrogen-containing compounds have been selected as efficient catalysts for carbon-carbon and carbonheteroatom forming reactions in a highly stereoselective fashion. Indeed, Bredig and Fiske have reported the use of cinchona alkaloid as the chiral organobase to accelerate enantioselective cyanohydrin synthesis early in 1912 [4]. However, chiral organobase catalysis did not have a wide application until much growing attention to organocatalysis nearly three decades ago. In this case, common chiral tertiary amines with weak basicity, for example, the use of cinchona alkaloid derivatives [5, 6] was surging. To match the variable basicity of catalysts for diverse organic reactions, chiral organosuperbases including guanidines [7-11], cyclopropenimines [12] and iminophosphazenes [13] were designed and synthesized in the past two decades (Fig. 1).

|
Download:
|
| Fig. 1. General classification of organobases. | |
{kind=link}
Chiral organobases are conceptually defined as a chiral organic molecular entity capable of abstracting protons. They are able to capture a proton from acidic pro-nucleophiles to generate a carbanion, and then the protonated catalysts afford strong molecular interaction with the carbanion through hydrogen bonding or electrostatic interaction. Moreover, chiral organobase catalysts which usually possess another functional group besides base site may demonstrate dual functionalizations, activating the pro-nucleophiles and electrophiles simultaneously.
According to the stability of conjugation system after protonation under reversible conditions, the basicity of an organobase could be identified. A direct way although not precise enough is to count the numbers of the canonical forms. Thus, the amine derivatives as amines, amidines, guanidines, cyclopropenimines and iminophosphazenes could be arranged in an alkaline order fromweak to strong (Fig. 1). In previous reports, several groups have commendably presented each kind of these chiral organobases. To the aim of introducing the highly topical area, herein, we will provide a brief summary of several representative kinds of chiral organobases and relatedapplications in asymmetricsynthesis.The chemistryof these organobases is full, but this review will not be comprehensive and several selected asymmetric catalytic reactions are presented to elucidate the catalytic characteristics.
2. Chiral tertiary aminesAmines are the widely used organic bases. Primary and secondary amines which could participate in organocatalysis via imine or enamine activation manner will be excluded herein. Diamines and other amine-based catalysts in combine with protonic acid could functionalize as hydrogen-bond donors are also out of the introduction. Tertiary amines including cinchona alkaloids and tertiary amine-based bifunctional organobasic catalysts are discussed in brief.
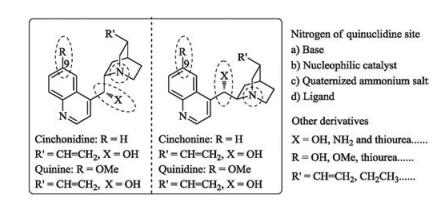
2.1. Cinchona alkaloids and their derivativesCinchona alkaloids, which were isolated from the bark of several species of cinchona trees in the early seventeenth century, are the earliest discovered and most representative tertiary amine core-containing chiral organobase (Fig. 2). The four basic structures usually refer as pseudo-enantiomeric forms (quinine and quinidine, cinchonine and cinchonidine). The nitrogen atom of the quinuclidine is generally viewed as the source of a base; and it also can be used as chiral nucleophilic catalyst. The first application of cinchona alkaloids in organic chemistry was used as chiral resolving reagents in 1853 [14]. Over the past 160 years, cinchona alkaloids and their derivatives undergo two renaissances. Nowadays, they have been developed into privileged catalysts for a variety of asymmetric reactions, such as asymmetric oxidations, asymmetric reductions, nucleophilic addition reactions and so on [5, 6].

|
Download:
|
| Fig. 2. Cinchona alkaloids and their derivatives. | |
{kind=link}
Cinchona alkaloids could be modified at several sites to achieve diverse bifunctional chiral skeletons. Chen's group has substituted the OH group into NH2 group, resulting in new bifunctional diamine catalyst 1a [15]. Further derivatizations, such as thiourea, squaramides, amides and so on, could act as hydrogen bonding donors.
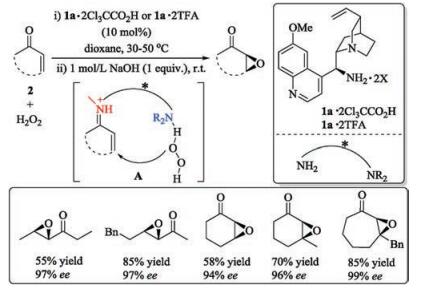
For example, catalytic asymmetric oxidation of olefins is one of the most important protocols to construct chiral epoxides. List's group utilized cinchona alkaloids derivative 1a to accelerate epoxidation of α, β-unsaturated ketones with hydrogen peroxide as the oxidant (Scheme 1) [16, 17]. They proposed a dual activation mode. The key iminium ion intermediate A may be formed by condensation between the primary amine site of the catalyst 1a and ketones 2, and the quinuclidine site of 1a may combine to hydrogen peroxide via hydrogen bonding. The corresponding chiral epoxides could be obtained in moderate to high yields with excellent ee values.

|
Download:
|
| Scheme 1. Cinchona alkaloid-derived diamine 1a catalyzed asymmetric epoxidation reaction of α, β-unsaturated ketones. | |
{kind=link}
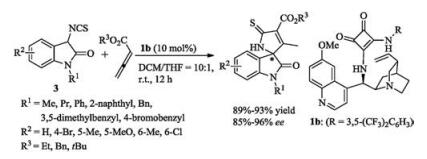
Cinchona alkaloid-derived thiourea 1b was employed by Shi and coworkers in a formal asymmetric [3 + 2]-cycloaddition of 3- isothiocyanatooxindoles 3 with allenic esters, affording different functionalized spirooxindole derivatives in good yields with high ee values under mild reaction conditions [18]. In this case, the quinuclidine site of the catalyst 1b abstracted one proton from oxindole 3 and the squaramide provided hydrogens to bind the allenic ester. Next, an intermolecular Michael addition/cyclization/ isomerization occurred to yield the product (Scheme 2).

|
Download:
|
| Scheme 2. Asymmetric [3 + 2]-cycloaddition reaction of 3-isothiocyanato-2- oxindoles with allenic esters catalyzed by cinchona-derived thiourea. | |
{kind=link}
Cinchona alkaloids and their derivatives have other rich application in asymmetric catalysis. The existence of nitrogen atom could also make cinchona alkaloids to be effective ligands for metal-promoted reactions, and one of the most famous examples is the osmium-catalyzed asymmetric dihydroxylation of olefins [19]. Alkylation of nitrogen atom giving chiral quaternized ammonium salts could be served as phase transfer catalysts.
2.2. Chiral diamine derivativesOptically active diamines, such as chiral 1, 10-binaphthyl-2, 20- diamine, 1, 2-diphenylethane-1, 2-diamine and cyclohexane-1, 2- diamine can be derived into valuable chiral tertiary amine-based bifunctional organocatalysts. Salient examples are shown in Fig. 3. Thioureas (4-6) and amide (7) subunits are introduced to functionalize as Brønsted acid units [20, 21].

|
Download:
|
| Fig. 3. Bifunctional organocatalysts containing tertiary amines. | |
{kind=link}
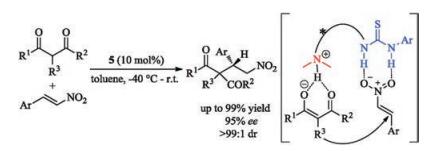
For instance, axially chiral catalyst 5 bearing a tertiary amine with a thiourea group is able to catalyze Michael addition of 1, 3- dicarbonyl compounds to trans-β-nitroalkenes with high enantioselectivities (Scheme 3) [21]. The tertiary amino group acts as an organobase to abstract a proton from the enolized 1, 3-dicarbonyl compounds, meanwhile the thiourea group activated the nitroalkenes via two hydrogen bonding. The conjugate addition took place enantioselectively to generate the corresponding products.

|
Download:
|
| Scheme 3. Tertiary diamine-derived catalyst 5 in Michael reaction of 1, 3- dicarbonyl compounds to nitrostyrenes. | |
{kind=link}
3. Chiral amidines
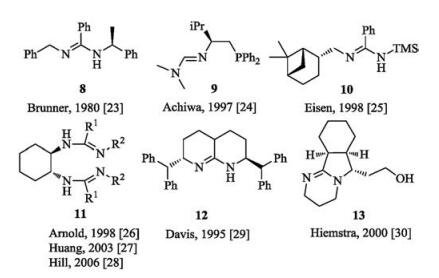
Amidines are a class of oxoacid derivatives with two nitrogen atoms in amino and imino groups of different functionalities [22]. An obvious feature of amidines is electron delocalization, leading to stronger basicity than usual amines (Fig. 4). Several kinds of chiral amidines were developed, which could be classified into two types: acyclic and cyclic ones (Fig. 5). Actually, most of them are used as ligands in metal complex involved reactions [23-29], and the applications of chiral amidines in asymmetric organocatalysis [30, 31] are rare due to the difficulty to achieve high enantioselectivity via modification of the amidine units.

|
Download:
|
| Fig. 4. General structures of amidines. | |
{kind=link}

|
Download:
|
| Fig. 5. Kinds of chiral acyclic and cyclic amidines. | |
{kind=link}
Intrigued by the bifunctional catalytic mode of cinchona alkaloids, Hiemstra and coworkers introduced amidine function as a stronger base in combination of a hydroxy group [30]. Hydroxy amidine 13 (Fig. 5) prepared from (S)-pyroglutamic acid could catalyze conjugate addition of thiophenol or dimethyl malonate to cyclohexenone, although the enantioselectivity-inducing properties are inferior to cinchona alkaloids.
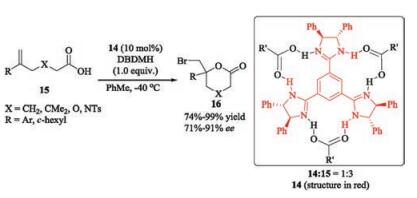
In 2010, Fujioka reported that C3-symmetric chiral trisimidazoline can catalyze asymmetric bromolactonization of δ, ε-unsaturated acids with 1, 3-dibromo-5, 5-dimethylhydantoin (DBDMH) as a brominating agent, giving bromolactones with high ee values [32]. The authors proposed that the catalyst 14 derived from 1, 2-diphenylethylenediamine can form a 1:3 complex with carboxylic acid 15 through hydrogen bonding (Scheme 4), which was supported by 1H NMR spectroscopic experiments. However, the possibility of the bifunctional catalyst 14 activated both 15 and DBDMH via hydrogen bonding cannot be ruled out.

|
Download:
|
| Scheme 4. Chiral amidines catalyzed asymmetric bromolactonization. | |
{kind=link}
4. Chiral guanidines
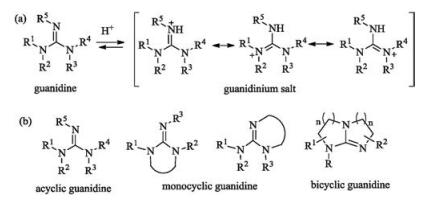
Guanidines (Fig. 6a) are organic superbases due to the high resonance stability of their conjugated protonated forms [7-11]. The presence of three nitrogen atoms bearing five possible chiral substituents may make them easy to be modified. Based on the guanidinyl group is incorporated into ring systems or not, they can be classified into three types, including acyclic, monocyclic and bicyclic ones (Fig. 6b).

|
Download:
|
| Fig. 6. (a) The conjugated protonated forms of guanidine. (b) Three typical structures of chiral guanidines. | |
{kind=link}
4.1. Chiral acyclic guanidines
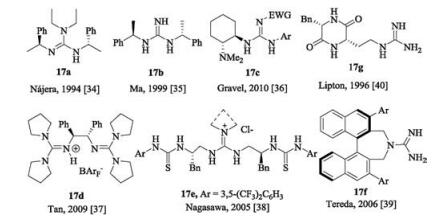
For chiral acyclic guanidines, a representative example is the amino acid of L-arginine in nature which contains an acyclic guanidyl terminal group. The classic synthetic acyclic guanidine developed by Barton in 1981 is called 'Barton's bases' [33]. Early chiral acyclic guanidine catalysts are listed in Fig. 7. The guanidine 17a [34] bears two ethyl substituents at the position of one nitrogen of amino group, presenting steric hindrance in comparison with 17b [35]. Early in 1994, Nájera and coworkers utilized chiral guanidine 17a to catalyze asymmetric Henry reaction of aldehydes with nitromethane albeit moderate yields and enantioselectivities were given [34]. Later, several acyclic guanidines and the corresponding salts are synthesized from chiral diamines, such as 17c by Gravel [36], 17d by Tan [37] and 17e by Nagasawa [38], respectively. Additionally, the Terada group prepared axially chiral acyclic guanidine 17f based on binaphthyl skeleton [39]. Lipton's group developed cyclic dipeptide-derived chiral catalyst 17g bearing a terminal guanidyl group [40]. These organocatalysts have been utilized in asymmetric Michael reaction, Mannich reaction and so on.

|
Download:
|
| Fig. 7. Salient examples of chiral acyclic guanidines. | |
{kind=link}
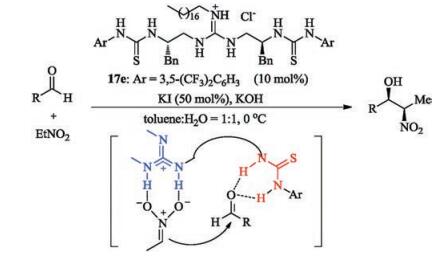
C2-Symmetric guanidine-thiourea catalyst 17e was developed to promote asymmetric Henry reaction of aldehydes with nitroethane [41]. It was found that the reaction performed well in the presence of guanidinium salt and KOH, yielding the nitropropanol derivatives in excellent yield as well as good diastereo- and enantio-selectivity (Scheme 5). The catalyst works as a dualactivation manner: the guanidine unit activates the nitroethane, and the thiourea actives the aldehyde through hydrogen-bonding.

|
Download:
|
| Scheme 5. Chiral guanidine-thiourea catalyzed asymmetric Henry reaction. | |
{kind=link}
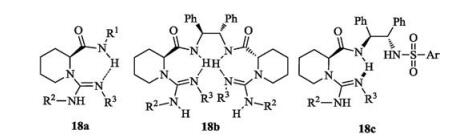
Based on the strategy of bifunctional catalysis, our group designed and developed chiral mono- or bis-guanidines from chiral amino acids and amines or diamines. Representative structures are listed in Fig. 8, including mono-guanidine-amides 18a, bisguanidines 18b and its hemi-salt, and guanidine- diamides 18c and other variation. The catalyst structures could be easily tuned by using variable amino acids and amines to meet the requirement of steric hindrance. These guanidines are useful bifunctional organocatalysts which have been utilized in several kinds of enantioselective transformations, such as inverse electron-demand hetero-Diels-Alder reaction [42], conjugate additions [43-46], aza-Mannich cascade reaction [47-50], oxyamination [51], Henry reaction [52] and α-hydroxylation [53] etc. [54, 55].

|
Download:
|
| Fig. 8. Chiral acyclic guanidines based on amino amide backbones. | |
{kind=link}
For instance, chiral monoguanidine 18a enabled the diastereoand enantio-selective Michael reaction between β-ketoesters and nitroolefins in excellent outcomes (Scheme 6) [56]. X-ray crystal diffraction analysis of guanidine-amide 18a showed that the catalyst formed one intramolecular hydrogen bonding between the amide skeleton and imine unit of guanidine, and one intermolecular hydrogen bonding between amidine unit and water. Thus, a dual-activation mode could be proposed. Guanidine unit functionalizes as a base to deprotonate the β-ketoester, and intramolecular H-bonding breaks, generating protonated guanidine unit to bond the nucleophile. On the other side, the released amide unit interacts with the nitroolefin as a H-bonding donor. The special environment created by the amino acid and amide in concert benefits the facial selectivity of the two reactants, giving satisfied diastereo- and enantio-selectivity.

|
Download:
|
| Scheme 6. Chiral guanidine-amide catalyzed Michael reaction of β-ketoesters with nitroolefins. | |
{kind=link}
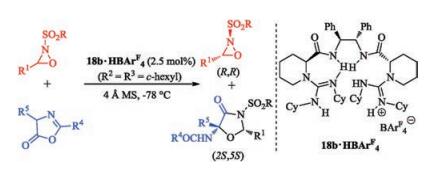
Recently, the HBAr4F salt of bisguanidines 18b was selected to promote the oxyamination of azlactones with oxaziridines (Scheme 7). Efficient catalytic asymmetric oxyamination and kinetic resolution were achieved simultaneously. We assumed that the salt 18b·HBAr4F acts bifunctional roles. The guanidine section of the catalyst may activate the azlactone and the guanidinium section preferentially recognize and activate the (S, S)-oxaziridine, giving a series of optically active oxazolin-4-one derivatives and oxaziridines [51].

|
Download:
|
| Scheme 7. Enantioselective oxyamination and kinetic resolution of oxaziridines catalyzed by chiral bisguanidine salt. | |
{kind=link}
4.2. Chiral monocyclic guanidines
In general, chiral monocyclic guanidines can be obtained by two methodologies. One is N-cyanation reaction of diamines with cyanogen bromide; and the other one is coupling reaction between 1, 3-disubstituted 2-iminoimidazolidines with or without diphenyl groups and primary amines (Fig. 9). Limited to the synthesis methodology, the chiral sources for the construction of monocyclic guanidines mainly come from diamines, phenylethylamine, naphthalene skeleton, and tartrate derivatives [35, 57-60]. Terada and coworkers designed both acyclic guanidine 17f and monocyclic guanidine 19f based on axially chiral binaphthyl skeleton [59]. Wang and Qu's group synthesized the corresponding guanidine 19g from tartaric acid [60]. These monocyclic guanidines themselves could work as bifunctional organocatalysts.

|
Download:
|
| Fig. 9. Two common synthetic protocols of chiral monocyclic guanidines and some selected examples. | |
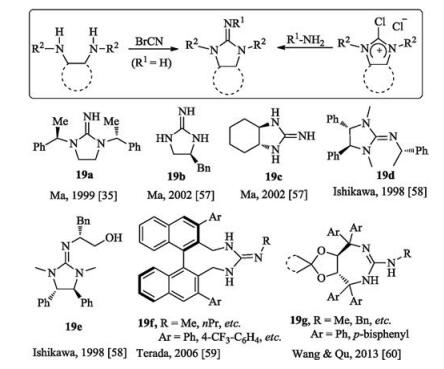{kind=link}
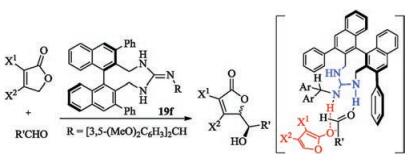
Enantioselective direct vinylogous aldol reaction of furanone derivatives catalyzed by the axially chiral guanidine base 19f was achieved by Terada and coworkers (Scheme 8) [61]. The high diastereo- and enantio-selectivity suggested the guanidinium ion, generated from the deprotonation of furanone derivatives would interact with both the anionic nucleophile and aldehyde through hydrogen-bonding interactions. The diastereoselectivity is dependent on the benzhydryl moiety adhered to the imine-nitrogen of the guanidine.

|
Download:
|
| Scheme 8. Chiral monocyclic guanidine catalyzed vinylogous aldol reaction of furanone derivatives. | |
{kind=link}
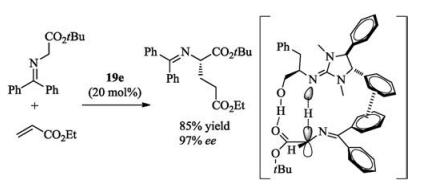
Ishikawa combined a hydroxy group into the guanidine 19d, and obtained a new multifunctional guanidine 19e catalyst [58]. In the presence of the catalyst 19e, Mannich reaction between ethyl acrylate and diphenyliminoglycinate took place efficiently (Scheme 9). In this case, a remarkable rate-acceleration was observed in the absence of solvents. A fixed complex of 19e with diphenyliminoglycinate with non-bond interactions as shown in Scheme 9 was rationalized to explain the enantioselectivity in the product [62].

|
Download:
|
| Scheme 9. Chiral monocyclic guanidine catalyzed Michael reaction. | |
{kind=link}
4.3. Chiral bicyclic guanidines
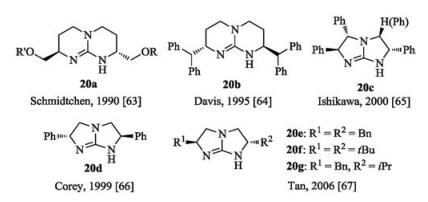
Chiral bicyclic guanidines generally include three frameworks as shown in Fig. 10. Schmidtchen and Davis reported the synthesis of TBD-based bicyclic guanidine 20a and 20b, respectively [63, 64]. Ishikawa and coworkers prepared tri- and tetra-substituted fivemembered bicyclic guanidine 20c [65]. The five-membered bicyclic chiral guanidines 20d-g were synthesized from the corresponding amino or aziridines by Corey's [66] and Tan's groups [67], respectively. These rigid bicyclic guanidines were found to be effective catalysts to promote base-catalyzed asymmetric reactions [68-74].

|
Download:
|
| Fig. 10. Structures of various chiral bicyclic guanidines. | |
{kind=link}
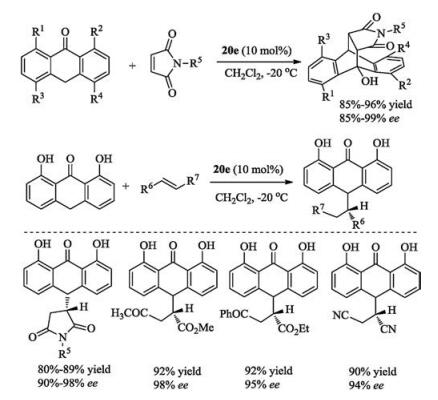
The Corey group utilized bicyclic guanidine 20d to accelerate the asymmetric Strecker reaction. This guanidine could activate both the aldimines and hydrocyanic acid simultaneously [66]. In 2006, the Tan group reported the chiral guanidine 20e-catalyzed Diels-Alder reaction of anthrones with pyrrole-2, 5-dione derivatives, and conjugate additions of anthrones with various Michael acceptors [75], affording divers desired adducts in good yields with high ee values (Scheme 10). These guanidines have also been extended to promote other reactions, such as enantioselective protonation, trimethylsilylcyanation reaction and so on [9, 10].

|
Download:
|
| Scheme 10. Chiral bicyclic guanidines catalyzed enantioselective Diels-Alder reaction and Michael addition of anthrones. | |
{kind=link}
For above chiral bicyclic guanidines, the corresponding guanidiniums can act as bifunctional catalysts to activate both the nucleophile and electrophile through hydrogen bonding. In 2011, Tan's group utilized chiral guanidine to catalyze decarboxylative Mannich reaction between malonic acid half thioesters 21 and imine (Scheme 11) [76]. Firstly, guanidine 20f abstracts the proton from malonic acid, and then the formed guanidium provides dual hydrogen bonding interactions with deprotonated malonic acid half thioesters and imine, simultaneously. Nucleophilic addition results in the formation of intermediate 23, and subsequent decarboxylation gives the final product. The DFT calculation and electrospray ionization (ESI) mass-spectrometric analysis supported this proposed pathway.

|
Download:
|
| Scheme 11. Chiral guanidine catalyzed enantioselective decarboxylative Mannich reaction. | |
{kind=link}
5. Chiral cyclopropenimines
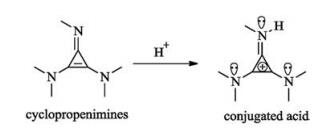
Cyclopropenimines contain one imino group and two amino groups at the position of three carbon atoms of cyclopropene. This new kind of organosuperbase that has stronger basicity than amidines and guanidines because their corresponding conjugated acid could be stabilized by the nitrogen lone electron pairs and aromatic cyclopropenium ion (Fig. 11).

|
Download:
|
| Fig. 11. Cyclopropenimines as strong organosuperbase. | |
{kind=link}
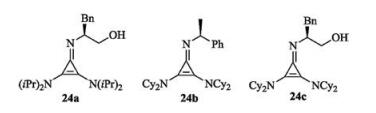
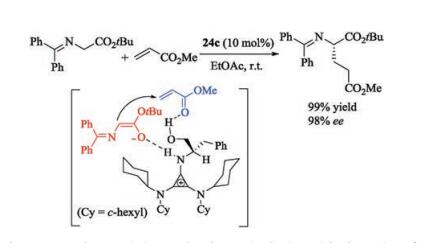
In 1971, Yoshida and Tawara reported the synthesis of chlorocyclopropenium salt [77]. On the basis of this work, Lambert and his coauthors designed chiral cyclopropenimines 24 by using (S)-2-amino-3-phenylpropan-1-ol or other chiral primary amine as the source of chirality (Fig. 12). In 2012, this group firstly reported the application of chiral cyclopropenimines 24c in asymmetric Michael reaction of glycine imine with methyl acrylate [78]. A dual activation mode was proposed in this work (Scheme 12). Initially, deprotonation of glycine imine with cyclopropenimine 24c, gives an ion pairing species. On the other hand, the catalyst bearing a hydroxyl activates methyl acrylate via hydrogen bonding. Conjugated attack results in the Michael adducts. In addition, the gram scale experiment demonstrated high efficiency of the chiral cyclopropenimines. The same group also applied the catalyst to asymmetric Mannich reaction of glycine imine with N-Boc-imines [79].

|
Download:
|
| Fig. 12. Chiral cyclopropenimines. | |
{kind=link}

|
Download:
|
| Scheme 12. Cyclopropenimine catalyzed enantioselective Michael reaction of glycine imine with methyl acrylate. | |
{kind=link}
6. Chiral iminophosphoranes
Iminophosphoranes, which were first prepared in 1973 [80], are identified as the strongest organobase around the found references. They can be generally classified into P1–P7 phosphazene bases based on the numbers of phosphorus atoms of iminophosphoranes (Fig. 13). Up to now, only chiral P1 iminophosphoranes has been synthesized and applied in base-catalyzed asymmetric reactions, and the other types of iminophosphoranes are limited to be used as achiral bases [1, 81-83]. For example, Bandar recently reported anti-Markovnikov alcohol addition reaction to aryl alkenes by using a kind of achiral iminophosphorane P4 catalyst [84].

|
Download:
|
| Fig. 13. General structures of representative iminophosphoranes. | |
{kind=link}
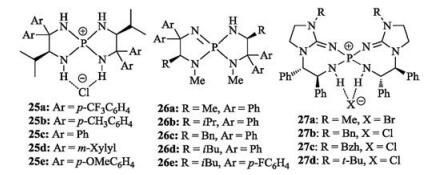
In 2007, Ooi's group reported a representative chiral P1-type iminophosphorane. A phosphorus-centered [5.5]-spirocyclic core was obtained through several steps from L-valine (Fig. 14) [85]. According to the single-crystal X-ray diffraction analysis of 25, it was found that chloride ion strongly binds to phosphorus cation via hydrogen bonding. The salts 25 are effective precursors of organobase. They can in situ generate organobase in the presence of catalytic loading of tBuOK, catalyzing Henry reaction of nitroalkane with aldehydes [85] to give β-hydroxyl nitroalkanes, or promoting Pudovik reaction of dimethyl H- phosphonate with aldehyde to yield α-hydroxyphosphonates in high yields with high ee values [86] (Scheme 13). Later, the same group synthesized organobase 26 existing in the form of iminophosphoranes, which can efficiently catalyze 1, 6- and 1, 8- Michael additions of azlactones to di- and trienyl N-acylpyrroles [87].

|
Download:
|
| Fig. 14. Structures of chiral phosphorus-centered [5.5]-spirocyclic and [7, 7]- spirocyclic iminophosphoranes. | |
{kind=link}

|
Download:
|
| Scheme 13. P-Spirocyclic iminophosphoranes catalyzed Henry reaction and Pudovik reaction. | |
{kind=link}
Terada designed and synthesized novel chiral [7, 7]-P-spirocyclic iminophosphoranes 27 in four steps (Fig. 14) [88-90]. The new chiral bis(guanidino)-iminophosphoranes possess two guanidine groups. Asymmetric amination of 2-alkyltetralones with azodicarboxylate was explored to validate synthetic potential of 27. It was found that less-acidic pro-nucleophiles could smoothly deprotonate to occur nucleophilic addition to azodicarboxylate in the presence of 27a and NaN(SiMe3)2, constructing a quaternary chiral center at the α-position of tetralone derivatives with high ee values (Scheme 14) [88].

|
Download:
|
| Scheme 14. Chiral bis(guanidino)-iminophosphorane catalyzed amination reaction. | |
{kind=link}
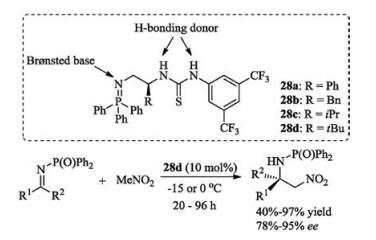
Dixon's group developed a new class of iminophosphoranes 28, which possess bifunctional Brønsted base/H-bonding donor [91-93]. The triphenylphosphine would create a strongly Brønsted basic site, and the thiourea group acts as a good hydrogen bonding donor. The bifunctional organobase 28d could successfully promote the challenging enantioselective organocatalytic azaHenry reaction of nitromethane with unactivated ketimines (Scheme 15) [91].

|
Download:
|
| Scheme 15. Iminophosphoranes catalyzed aza-Henry reaction of ketimine. | |
{kind=link}
7. Summary and outlook
Over the past decades, chiral organobases attract a great of attention because of their unique features and wide application in organic chemistry. These chiral organobases generally include chiral tertiary amines, amidines, guanidines, cyclopropenimines and iminophosphoranes. Among them, chiral guanidines, cyclopropenimines and iminophosphoranes are organosuperbases, which were designed and synthesized by various groups in recent twenty years. Chiral organobases can be obtained from natural products or readily available chiral amino acids and diamines. With or without the assistance of additional functional groups, these organocatalysts enable a number of base-initiated asymmetric reactions. Moreover, a bifunctional catalytic fashion via hydrogenbonding or other weak interactions between the reactants with the catalyst subunits guarantee high stereoselectivities of the reactions. Nonetheless, the level of the development of chiral organobases is still limited so far. Most organobases-catalyzed reactions are constrained to use acidic pronucleophile substrates. In addition, the sources of chiral scaffolds are also limited to chiral nitrogen-containing compounds. Thus, new types of chiral organobases and different kinds of substrates are anticipated in future development.
AcknowledgmentsWe thank the National Natural Science Foundation of China (Nos. 21625205 and 21332003), and the National Program for Support of Top-Notch Young Professionals for financial support.
| [1] |
T. Ishikawa, Superbases for Organic Synthesis, Wiley, Chippenham, 2009.
|
| [2] |
C. Palomo, M. Oiarbide, R. López, Chem. Soc. Rev. 38 (2009) 632-653. DOI:10.1039/B708453F |
| [3] |
J.E. Taylor, S.D. Bull, J.M.J. Williams, Chem. Soc. Rev. 41 (2012) 2109-2121. DOI:10.1039/c2cs15288f |
| [4] |
G. Bredig, P.S. Fiske, Biochem. Z. 46 (1912) 7-23. |
| [5] |
C. E. Song, Cinchona Alkaloids in Synthesis and Catalysis, Wiley-VCH, Weinheim, 2009.
|
| [6] |
G.S. Singh, E.M. Yeboah, Rep. Org. Chem. 6 (2016) 47-75. DOI:10.2147/ROC |
| [7] |
T. Ishikawa, T. Isobe, Chem. -Eur. J. 8 (2002) 552-557. DOI:10.1002/(ISSN)1521-3765 |
| [8] |
K. Nagasawa, Y. Hashimoto, Chem. Rec. 3 (2003) 201-211. DOI:10.1002/(ISSN)1528-0691 |
| [9] |
D. Leow, C. H. Tan, Chem. Asian J. 4 (2009) 488-507. DOI:10.1002/asia.v4:4 |
| [10] |
X. Fu, C. H. Tan, Chem. Commun. 47 (2011) 8210-8222. DOI:10.1039/c0cc03691a |
| [11] |
C. Alonso-Moreno, A. Antiñolo, F. Carrillo-Hermosilla, A. Otero, Chem. Soc. Rev. 43 (2014) 3406-3425. DOI:10.1039/C4CS00013G |
| [12] |
J.S. Bandar, T.H. Lambert, Synthesis 45 (2013) 2485-2498. DOI:10.1055/s-00000084 |
| [13] |
H. Krawczyk, M. Dzie, gielewski, D. Deredas, A. Albrecht, Ł. Albrecht, Chem.-Eur. J. 21 (2015) 10268-10277. DOI:10.1002/chem.201500481 |
| [14] |
L. Pasteur, Acad. Sci. 37 (1853) 162-166. |
| [15] |
L. Jiang, Y. C. Chen, Catal. Sci. Technol. 1 (2011) 354-365. DOI:10.1039/c0cy00096e |
| [16] |
C.M. Reisinger, X. Wang, B. List, Angew. Chem. Int. Ed. 47 (2008) 8112-8115. DOI:10.1002/anie.v47:42 |
| [17] |
X. Wang, C.M. Reisinger, B. List, J. Am. Chem. Soc. 130 (2008) 6070-6071. DOI:10.1021/ja801181u |
| [18] |
D. Du, Y. Jiang, Q. Xu, M. Shi, Adv. Synth. Catal. 355 (2013) 2249-2256. DOI:10.1002/adsc.201300460 |
| [19] |
H.C. Kolb, M.S. VanNieuwenhze, K.B. Sharpless, Chem. Rev. 94 (1994) 2483-2547. DOI:10.1021/cr00032a009 |
| [20] |
J. Wang, H. Li, W. Duan, L. Zu, W. Wang, Org. Lett. 7 (2005) 4713-4716. DOI:10.1021/ol0519137 |
| [21] |
T. Okino, Y. Hoashi, T. Furukawa, X. Xu, Y. Takemoto, J. Am. Chem. Soc. 127 (2005) 119-125. DOI:10.1021/ja044370p |
| [22] |
F. P. Camponovo, Chiral Ferrocenyl Amidines as Modular Ligands for Applications in Asymmetric Catalysis, ETH, Zürich, 2009.
|
| [23] |
H. Brunner, G. Agrifoglio, I. Bernal, M.W. Creswick, Angew. Chem. Int. Ed. 19 (1980) 641-642. DOI:10.1002/(ISSN)1521-3773 |
| [24] |
A. Saitoh, T. Morimoto, K. Achiwa, Tetrahedron:Asymmetry 8 (1997) 3567-3570. DOI:10.1016/S0957-4166(97)00482-5 |
| [25] |
C. Averbuj, E. Tish, M.S. Eisen, J. Am. Chem. Soc. 120 (1998) 8640-8646. DOI:10.1021/ja980392y |
| [26] |
J.R. Hagadorn, J. Arnold, Angew. Chem. Int. Ed. 37 (1998) 1729-1731. DOI:10.1002/(ISSN)1521-3773 |
| [27] |
J.F. Li, S.P. Huang, L.H. Weng, D.S. Liu, Eur. J. Inorg. Chem. (2003) 810-813. |
| [28] |
M.S. Hill, P.B. Hitchcock, S.M. Mansell, Dalton Trans. (2006) 1544-1553. |
| [29] |
M.A. Convery, A.P. Davis, C.J. Dunne, J.W. MacKinnon, Tetrahedron Lett. 36 (1995) 4279-4282. DOI:10.1016/0040-4039(95)00739-Y |
| [30] |
M. Ostendorf, S. van der Neut, F.P.J.T. Rutjes, H. Hiemstra, Eur. J. Org. Chem. (2000) 105-113. |
| [31] |
M. Ostendorf, J. Dijkink, F.P.J.T. Rutjes, H. Hiemstra, Eur. J. Org. Chem. (2000) 115-124. |
| [32] |
K. Murai, T. Matsushita, A. Nakamura, S. Fukushima, M. Shimura, H. Fujioka, Angew. Chem. Int. Ed. 49 (2010) 9174-9177. DOI:10.1002/anie.201005409 |
| [33] |
D.H.R. Barton, J.D. Elliott, S.D. GÉro, J. Chem. Soc. Chem. Commun. (1981) 1136-1137. |
| [34] |
R. Chinchilla, C. Nájera, P. Sánchez-Agulló, Tetrahedron:Asymmetry 5 (1994) 1393-1402. DOI:10.1016/0957-4166(94)80183-5 |
| [35] |
D. Ma, K. Cheng, Tetrahedron:Asymmetry 10 (1999) 713-719. DOI:10.1016/S0957-4166(99)00038-5 |
| [36] |
K. Thai, M. Gravel, Tetrahedron:Asymmetry 21 (2010) 751-755. DOI:10.1016/j.tetasy.2010.04.033 |
| [37] |
X. Fu, W.T. Loh, Y. Zhang, et al., Angew. Chem. Int. Ed. 48 (2009) 7387-7390. DOI:10.1002/anie.v48:40 |
| [38] |
Y. Sohtome, Y. Hashimoto, K. Nagasawa, Adv. Synth. Catal. 347 (2005) 1643-1664. DOI:10.1002/(ISSN)1615-4169 |
| [39] |
M. Terada, M. Nakano, H. Ube, J. Am. Chem. Soc. 128 (2006) 16044-16045. DOI:10.1021/ja066808m |
| [40] |
M.S. Iyer, K.M. Gigstad, N.D. Namdev, M. Lipton, J. Am. Chem. Soc. 118 (1996) 4910-4911. DOI:10.1021/ja952686e |
| [41] |
K. Takada, N. Takemura, K. Cho, Y. Sohtome, K. Nagasawa, Tetrahedron Lett. 49 (2008) 1623-1626. DOI:10.1016/j.tetlet.2008.01.030 |
| [42] |
S.X. Dong, X.H. Liu, X.H. Chen, et al., J. Am. Chem. Soc. 132 (2010) 10650-10651. DOI:10.1021/ja1046928 |
| [43] |
Y. Yang, S.X. Dong, X.H. Liu, L.L. Lin, X.M. Feng, Chem. Commun. 48 (2012) 5040-5042. DOI:10.1039/c2cc31470c |
| [44] |
X. Xiao, X.H. Liu, S.X. Dong, et al., Chem.-Eur. J. 18 (2012) 15922-15926. DOI:10.1002/chem.v18.50 |
| [45] |
Y.S. Chen, X.H. Liu, W.W. Luo, L.L. Lin, X.M. Feng, Synlett 28 (2017) 966-969. DOI:10.1055/s-0036-1588940 |
| [46] |
S. Ruan, X.B. Lin, L.H. Xie, et al., Org. Chem. Front. 5 (2018) 32-35. DOI:10.1039/C7QO00768J |
| [47] |
S.X. Dong, X.H. Liu, Y.L. Zhang, L.L. Lin, X.M. Feng, Org. Lett. 13 (2011) 5060-5063. DOI:10.1021/ol2018888 |
| [48] |
X.H. Chen, S.X. Dong, Z. Qiao, et al., Chem.-Eur. J. 17 (2011) 2583-2586. DOI:10.1002/chem.201002571 |
| [49] |
K.R. Yu, X.H. Liu, X.B. Lin, L.L. Lin, X.M. Feng, Chem. Commun. (2015) 14897-14900. |
| [50] |
S.S. Guo, X.H. Liu, B. Shen, L.L. Lin, X.M. Feng, Org. Lett. 18 (2016) 5070-5073. DOI:10.1021/acs.orglett.6b02522 |
| [51] |
S.X. Dong, X.H. Liu, Y. Zhu, et al., J. Am. Chem. Soc. 135 (2013) 10026-10029. DOI:10.1021/ja404379n |
| [52] |
B. Fang, X.H. Liu, J.N. Zhao, et al., J. Org. Chem. 80 (2015) 3332-3338. DOI:10.1021/acs.joc.5b00075 |
| [53] |
X.B. Lin, S. Ruan, Q. Yao, et al., Org. Lett. 18 (2016) 3602-3605. DOI:10.1021/acs.orglett.6b01614 |
| [54] |
F. Tan, X.H. Liu, X.Y. Hao, et al., ACS Catal. 6 (2016) 6930-6934. DOI:10.1021/acscatal.6b02184 |
| [55] |
Q. Zhang, S.S. Guo, J. Yang, et al., Org. Lett. 19 (2017) 5826-5829. DOI:10.1021/acs.orglett.7b02772 |
| [56] |
Z.P. Yu, X.H. Liu, L. Zhou, L.L. Lin, X.M. Feng, Angew. Chem. Int. Ed. 48 (2009) 5195-5198. DOI:10.1002/anie.v48:28 |
| [57] |
D. Ma, Q. Panb, F. Hana, Tetrahedron Lett. 43 (2002) 9401-9403. DOI:10.1016/S0040-4039(02)02332-8 |
| [58] |
T. Isobe, K. Fukuda, T. Ishikawa, Tetrahedron:Asymmetry 9 (1998) 1729-1735. DOI:10.1016/S0957-4166(98)00152-9 |
| [59] |
M. Terada, H. Ube, Y. Yaguchi, J. Am. Chem. Soc. 128 (2006) 1454-1455. DOI:10.1021/ja057848d |
| [60] |
L. Zou, B. Wang, H. Mu, H. Zhang, Y. Song, J. Qu, Org. Lett. 15 (2013) 3106-3109. DOI:10.1021/ol401306h |
| [61] |
H. Ube, N. Shimada, M. Terada, Angew. Chem. Int. Ed. 49 (2010) 1858-1861. DOI:10.1002/anie.v49:10 |
| [62] |
T. Ishikawa, Y. Araki, T. Kumamoto, et al., Chem. Commun. (2001) 245-246. |
| [63] |
H. Kurzmeier, F.P. Schmidtchen, J. Org. Chem. 55 (1990) 3749-3755. DOI:10.1021/jo00299a013 |
| [64] |
A.P. Davis, K.J. Dempsey, Tetrahedron:Asymmetry 6 (1995) 2829-2840. DOI:10.1016/0957-4166(95)00374-X |
| [65] |
T. Isobe, K. Fukuda, K. Yamaguchi, et al., J. Org. Chem. 65 (2000) 7779-7785. DOI:10.1021/jo000746f |
| [66] |
E.J. Corey, M.J. Grogan, Org. Lett. 1 (1999) 157-160. DOI:10.1021/ol990623l |
| [67] |
W. Ye, D. Leow, S.L.M. Goh, et al., Tetrahedron Lett. 47 (2006) 1007-1010. DOI:10.1016/j.tetlet.2005.11.133 |
| [68] |
V. Alcázar, J.R. Morán, J. de Mendoza, Tetrahedron Lett. 36 (1995) 3941-3944. DOI:10.1016/0040-4039(95)00647-U |
| [69] |
X. Fu, Z. Jiang, C.H. Tan, Chem. Commun. (2007) 5058-5060. |
| [70] |
Z. Jiang, W. Ye, Y. Yang, C.H. Tan, Adv. Synth. Catal. 350 (2008) 2345-2351. DOI:10.1002/adsc.v350:14/15 |
| [71] |
W. Ye, Z. Jiang, Y. Zhao, et al., Adv. Synth. Catal. 349 (2007) 2454-2458. DOI:10.1002/(ISSN)1615-4169 |
| [72] |
D. Leow, S. Lin, S.K. Chittimalla, X. Fu, C.H. Tan, Angew. Chem. Int. Ed. 47 (2008) 5641-5645. DOI:10.1002/anie.v47:30 |
| [73] |
Z. Jiang, Y. Pan, Y. Zhao, et al., Angew. Chem. Int. Ed. 48 (2009) 3627-3631. DOI:10.1002/anie.v48:20 |
| [74] |
J. Wang, J. Chen, C.W. Kee, C.H. Tan, Angew. Chem. Int. Ed. 51 (2012) 2382-2386. DOI:10.1002/anie.v51.10 |
| [75] |
J. Shen, T.T. Nguyen, Y.P. Goh, et al., J. Am. Chem. Soc. 128 (2006) 13692-13693. DOI:10.1021/ja064636n |
| [76] |
Y. Pan, C. Kee, Z. Jiang, et al., Chem.-Eur. J. 17 (2011) 8363-8370. DOI:10.1002/chem.201100687 |
| [77] |
Z. Yoshida, Y. Tawara, J. Am. Chem. Soc. 93 (1971) 2573-2574. DOI:10.1021/ja00739a057 |
| [78] |
J.S. Bandar, T.H. Lambert, J. Am. Chem. Soc. 134 (2012) 5552-5555. DOI:10.1021/ja3015764 |
| [79] |
J.S. Bandar, T.H. Lambert, J. Am. Chem. Soc. 135 (2013) 11799-11802. DOI:10.1021/ja407277a |
| [80] |
K. Issleib, M. Lischewski, Synth. React. Inorg. Met. Org. Chem. 3 (1973) 255-266. DOI:10.1080/00945717308057582 |
| [81] |
Z. Jin, S.H. Kim, P.L. Fuchs, Tetrahedron Lett. 37 (1996) 5247-5248. DOI:10.1016/0040-4039(96)01110-0 |
| [82] |
R. Schwesinger, H. Schlemper, Angew. Chem. Int. Ed. 26 (1987) 1167-1169. DOI:10.1002/(ISSN)1521-3773 |
| [83] |
W. Maier, M. Keller, W. Eberbach, Heterocycles 35 (1993) 817-820. DOI:10.3987/COM-92-S(T)64 |
| [84] |
C. Luo, J.S. Bandar, J. Am. Chem. Soc. 140 (2018) 3547-3550. DOI:10.1021/jacs.8b00766 |
| [85] |
D. Uraguchi, S. Sakaki, T. Ooi, J. Am. Chem. Soc. 129 (2007) 12392-12393. DOI:10.1021/ja075152+ |
| [86] |
D. Uraguchi, T. Ito, T. Ooi, J. Am. Chem. Soc. 131 (2009) 3836-3837. DOI:10.1021/ja810043d |
| [87] |
D. Uraguchi, K. Yoshioka, Y. Ueki, T. Ooi, J. Am. Chem. Soc. 134 (2012) 19370-19373. DOI:10.1021/ja310209g |
| [88] |
T. Takeda, M. Terada, J. Am. Chem. Soc. 135 (2013) 15306-15309. DOI:10.1021/ja408296h |
| [89] |
Q. Hu, A. Kondoh, M. Terada, Chem. Sci. 9 (2018) 4348-4351. DOI:10.1039/C8SC00808F |
| [90] |
A. Kondoh, S. Akahira, M. Oishi, M. Terada, Angew. Chem. Int. Ed. 57 (2018) 6299-6303. DOI:10.1002/anie.v57.21 |
| [91] |
M.G. Núñez, A.J.M. Farley, D.J. Dixon, J. Am. Chem. Soc. 135 (2013) 16348-16351. DOI:10.1021/ja409121s |
| [92] |
A.J.M. Farley, C. Sandford, D.J. Dixon, J. Am. Chem. Soc. 137 (2015) 15992-15995. DOI:10.1021/jacs.5b10226 |
| [93] |
J. Yang, A.J.M. Farley, D.J. Dixon, Chem. Sci. 8 (2017) 606-610. DOI:10.1039/C6SC02878K |