 2018, Vol. 29
2018, Vol. 29 


 ,
Wenjie Wua,
Zhangyong Hongb
,
Wenjie Wua,
Zhangyong Hongb
b State Key Laboratory of Medicinal Chemical Biology, Tianjin Key Laboratory of Protein Sciences, College of Life Sciences, Nankai University, Tianjin 300071, China;
c Department of General Surgery, Second Xiangya Hospital, Central South University, Changsha 410011, China
Photodynamic therapy (PDT) has emerged as an important noninvasive therapeutic modality in the management of a variety of premalignant and malignant diseases [1]. However, the limited availability of ideal photosensitizers with desired properties continues to be an important bottleneck in PDT therapy [2]. In recent decades, substantial effort has been made toward the development of new photosensitizers for PDT [3]. Among them, phthalocyanine (Pc) is one of the most promising candidates [4]. Pc has advantageous photophysical and photochemical properties, including strong absorption (extinction coefficient ε > 105 L mol-1 cm-1) at long wavelengths ( > 670 nm) and strong singlet oxygen generation abilities (a singlet oxygen quantum yield of approximately 50%) [5]. However, most Pcs exhibit extremely low solubility and strong tendency to aggregate in water due to the hydrophobic nature of the planar Pc macrocycle structure [6], which always renders Pcs photodynamically inactive in aqueous media [7] and significantly restricts their in vivo biological and medical applications. Many studies have reported on the chemical modification of Pcs through the attachment of hydrophilic substituents to peripheral positions on the macrocycle to increase the water solubility of Pcs [8, 9]. However, hydrophilic modification greatly reduces the PDT activity of Pcs, possibly by reducing their cell permeability and membrane attachment [3b].
Conjugating Pcs to peptide ligands could be another practical and useful strategy for improving the physical properties of Pcs [10, 11]. Conjugation with proper hydrophilic tumor-homing peptide ligands can increase water solubility and reduce the aggregation of Pcs. It is also possible to improve Pcs' ability to target tumors through interactions between receptors and the peptide ligands [12]. However, most studies have used nonmodified or alkyl-group-modified Pcs with which to conjugate peptide ligands. In this context, the problems associated with the strong hydrophobicity of Pcs, such as aggregation and low water solubility, cannot be well resolved by peptide conjugation. In addition, the strong hydrophobicity of Pcs enhances non-specific interactions with the cell membrane and destroys the binding function of the peptide ligands, increasing the non-specific background signal.
In our previous studies [13], using hydrophilic group-modified Pcs (i.e., modification with triethyleneglycol monomethyl ether substitution or glycol chain substitution) to construct peptide-Pc conjugates, the conjugates were well-shielded from non-specific binding with cells and showed improved tumor selectivity in cellbased experiments. However, the PDT efficiency obtained in the cell-based experiments was appreciably reduced after making these modifications, particularly for the relatively highly hydrophilic modification (glycol chain substitution). Compared with the relatively less hydrophilic modification, the highly hydrophilic modification appears to increase tumor selectivity and reduce the background distribution in vivo but with relatively low PDT efficiency. This disparity may require us to finely tune the hydrophilicity of peptide-conjugated Pcs to obtain a better PDT outcome. Moreover, the PDT potential of such Pcs for in vivo tumor treatment has not been tested in animal models. Thus, we set out to further optimize the structure of peptide-conjugated Pcs by varying the hydrophilic substitutions on the Pc ring and by adding a hydrophilic linker to finely tune the hydrophilicity, with the aim of balancing the tumor selectivity and PDT activity of the conjugates.
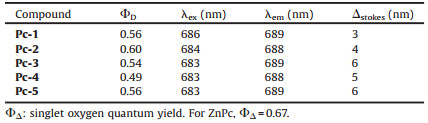
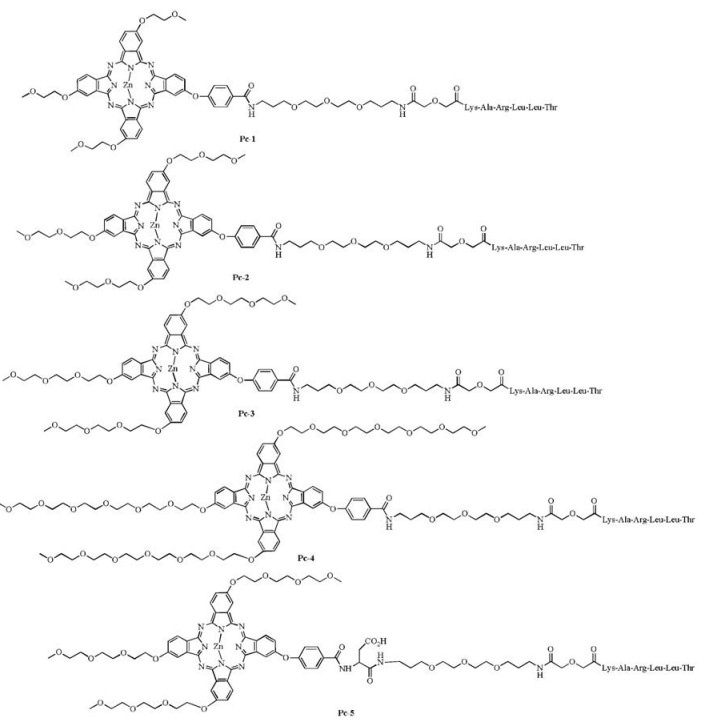
We designed a solid-phase strategy [14] for the synthesis of the proposed conjugates. For comparison, four hydrophilic substitutions (ethylene glycol monomethyl ether group, diethylene glycol monomethyl ether group, triethylene glycol monomethyl ether group and hexaethylene glycol monomethyl ether group) were adopted to reduce the hydrophobicity of the Pc ring component (Pc-1-4 in Fig. 1). Asymmetrically substituted A3B-type Pcs with hydrophilic substitutions on the peripheral positions of the macrocycle and a single carboxylic acid were designed to facilitate convenient conjugation with amine-containing peptide ligands. An extra polyethylene glycol (PEG) chain [15] and an extra glutamic residue (Pc-5 in Fig. 1) were incorporated between the Pc macrocycle component and peptide ligand to test their effect in further increasing water solubility of the conjugates and reducing the influence of the Pc aromatic macrocycle on the affinity of the peptide ligands. The short peptide Lys-Ala-Arg-Leu-Leu-Thr, a modified EGF receptor-targeting peptide [13b, 16], was coupled to Pcs for tumor-targeting purposes. Pc-Control (in Fig. 2), which contained the LLKATR ligand (a disorderly sequence of the EGFR targeting ligand KARLLT with no affinity to EGFR receptor), was synthesized as a control.

|
Download:
|
| Fig. 1. Design of EGFR targeting peptide-conjugated Pcs. Pc-1-4 feature ethylene glycol chains of varying lengths on the PC ring component. Pc-5 features an extra aspartic acid residue on the linker compared with Pc-3. | |

|
Download:
|
| Fig. 2. Structure of Pc-Control, with a disorderly peptide sequence and no EGFR targeting capability compared with Pc-3. | |
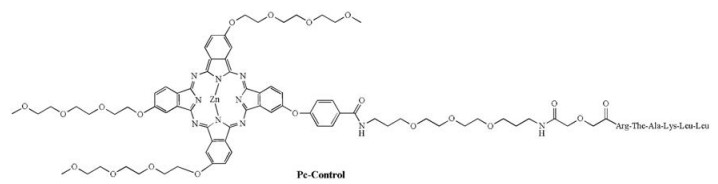
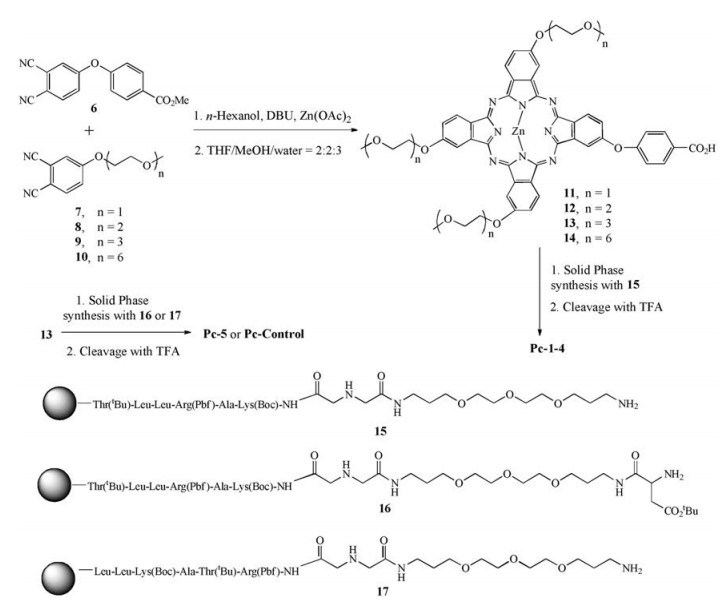
The synthetic route to the above mentioned Pc conjugates is shown in Scheme 1. Phthalonitrile 6 was submitted to statistical condensation with phthalonitrile 7, 8, 9 or 10 in n-hexanol at 160 ℃ for 7 h in the presence of Zn(Ⅱ) acetate and 1, 8-diazabicyclo[5.4.0] undec-7-ene (DBU), followed by hydrolysis with NaOH in THF/water/methanol (2/1/5) and subsequent acidification. The reaction afforded Pc 11, 12, 13 and 14 with a free carboxylic group for peptide conjugation. The yields for these Pcs typically ranged from 15% to 30% in two steps. The peptide NH2-Linker-Lys(Boc)-Ala-Arg (Pbf)-Leu-Leu-Thr(tBu)-resin 15 with protected side chains was prepared on a resin using a Fmoc solid-phase peptide synthesis (SPPS) protocol. The N terminus of peptide 15 was coupled to the carboxyl group of Pc 11, 12, 13 or 14 using 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) and 1-hydroxy-benzotriazole (HOBT) as the activating agents. The peptide-Pc conjugate was cleaved from the resin by removing all protecting groups at the same time in 95% TFA with triisopropylsilane (TIS) (TFA:H2O:TIS = 95:2.5:2.5) to obtain the desired conjugates Pc-1, Pc-2, Pc-3 and Pc-4 after HPLC purification. Similarly, Pc-5 and Pc-Control were prepared by coupling Pc 13 with side chainprotected NH2-Glu(tBu)-Linker-Lys(Boc)-Ala-Arg(Pbf)-Leu-LeuThr(tBu)-resin (16) or sequence-disordered NH2-Glu(tBu)-LinkerArg(Pbf)-Thr(tBu)-Ala-Lys(Boc)-Leu-Leu-resin (17) via solid-phase synthesis, followed by deprotection in 95% TFA (TFA:H2O:TIS = 95:2.5:2.5) in good yield after HPLC purification.

|
Download:
|
| Scheme 1. Synthesis of the designed peptide conjugated Pcs. | |
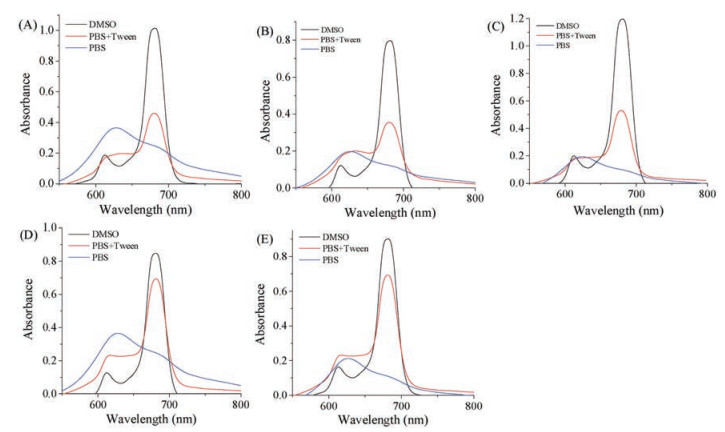
The UV–vis absorption spectra (Fig. 3 and Table 1) of the synthesized Pc conjugates were obtained in solvents, DMSO, PBS, or PBS containing a low concentration of lipids. In DMSO, the electronic spectra of all five synthesized Pc conjugates showed intense absorption in the Q band region at 680–681 nm, which is redshifted by approximately 10 nm compared with the Q band region of nonsubstituted PcZn (~670 nm). The conjugates also showed almost completely monomeric behavior, as indicated by a single, narrow Q band. All conjugates exhibited excellent solubility in aqueous solution, while they formed cofacial aggregates in PBS, as evidenced by the higher-energy (blue shifted) band at 620–640 nm in the Q band region. With the addition of 0.05% Tween 80 to the aqueous solution, the Q band at l = 600–700 nm became much sharper, and the spectral shape became similar to that in DMSO. This finding indicated that the addition of a very low concentration of lipids can effectively break up the aggregates in aqueous solution. The aggregation of Pc-4 with hexaglycol substitution and that of Pc-5 with an extra glutamic acid residue were relatively more easily disrupted, as demonstrated by the relatively sharper and stronger peak observed at 680 nm. The hydrophilic ethylene glycol chain surrounding the Pc rings, hydrophilic peptide ligand and hydrophilic PEG linker significantly contributed to relieving the aggregation problem associated with zinc Pcs.

|
Download:
|
| Fig. 3. UV–vis absorption spectra of Pc-1 (A), Pc-2 (B), Pc-3 (C), Pc-4 (D) and Pc-5 (E) in various solvents (i.e., DMSO, 0.05% Tween 80 in PBS and PBS solution). The concentration for absorption determination was 10 μmol/L. | |
|
|
Table 1 Absorption spectra data of Pc-1-5 in DMSO, 0.05% Tween 80 in PBS and PBS. Data are expressed as λmax/nm (log ε). |

Fluorescence spectra of the Pc conjugates were obtained in DMSO (Table 2 and Fig. S3 in Supporting information). All synthesized Pcs displayed a fluorescence excitation band at approximately 685 nm and an emission band at approximately 695 nm, which are typical for metal Pc complexes. The singlet oxygen quantum yields of these conjugates were determined in DMSO, as shown in Table 2. All five Pc conjugates showed roughly the same singlet oxygen quantum yield, in the range 0.49–0.60, comparable to that of non-substituted ZnPc (approximately 0.67). This finding indicates that peptide conjugation and Pc ring modification have only a very moderate effect on singlet oxygen quantum yield. Singlet oxygen (1O2) is believed to be the major cytotoxic agent involved in PDT, and a high singlet oxygen quantum yield should be highly important for photodynamic therapy (Table 3).
|
|
Table 2 Photophysical and photochemical parameters of Pc-1-5 in DMSO. |
|
|
Table 3 Phototoxicity of Pc conjugates determined by MTT assay. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
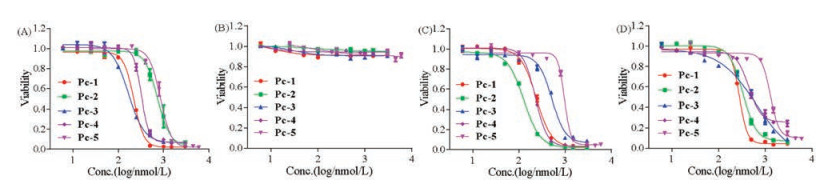
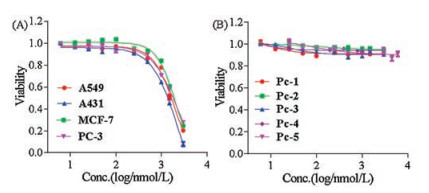
The in vitro PDT activity of Pc-1-5 was evaluated by measuring cellular survival rates in human tumor cell lines, including A431 and A549 cells, which express very high levels of EGFR, and MCF-7 and PC-3 cells, but exhibit relatively low expression levels of EGFR, via the MTT assay (Fig. 4). The results show that all five Pc conjugates exhibited substantial cytotoxicity when treated by irradiation for 10 min with a 660-nm laser. Among the conjugates, Pc-3 displayed the highest PDT activity and selectivity. Lower EC50 (50% inhibition of cell proliferation based on dose-response curves) levels were observed for A431 and A549 cells, approximately 220 nmol/L and 170 nmol/L, respectively, than for MC-7 and PC-3 cells, approximately 520 nmol/L and 650 nmol/L, respectively. Interestingly, Pc-5 showed an EC50 value of approximately 1000-1200 nmol/L towards these tumor cells, much higher than those of Pc-3. An extra carboxylic acid group in the linker appears to significantly reduce the PDT activity of the Pc conjugates. The negative charge of the carboxylic group may reduce the association of the conjugate towards the plasma or organellar membrane, thus reducing PDT activity. When the cells were treated with the conjugate Pc-Control—prepared by conjugation of Pc 13 with a disorderly sequence of EGFR peptide and no affinity toward EGFR receptor—we found much lower PDT activity towards these tumor cells, with EC50 values of 1900, 2100, 2400 and 2200 nmol/L for A431, A549, MCF-7 and PC-3 cells, respectively (Fig. 5A). The PDT activities towards A431 and A549 cells expressing high levels of EGFR receptor were reduced approximately ten-fold compared with the PDT activity of Pc-3 (1900 nmol/L, 2100 nmol/L ∝ 220 nmol/L, 170 nmol/L, respectively). These results indicate that peptide targeting is highly important for the PDT activity of these Pc conjugates. All conjugates were essentially non-cytotoxic in the dark toward all cell lines (Fig. 5B), and a cell viability of 90% was observed without irradiation after incubation with these conjugates at concentrations of up to 3000 nmol/L, indicating that these conjugates show very good biocompatibilities without exposure to irradiation.

|
Download:
|
| Fig. 4. Comparative in vitro phototoxicity efficacy of Pc-1, Pc-2, Pc-3, Pc-4 and Pc-5 toward A549 (A), A431 (B), MCF-7 (C) and PC-3 (D) cells (n = 5, mean ± SEM). Phototoxicity efficacy after cells were treated with a series of concentrations of Pc-1, Pc-2, Pc-3, Pc-4 and Pc-5 under illumination for 10 min (laser wavelength of 660 nm and laser power density of 40 mW/cm2). | |
{kind=link}

|
Download:
|
| Fig. 5. (A) Phototoxicity efficacy of Pc-Control, with disorderly peptide sequence, toward A549, A431, MCF-7 and PC-3 cells after cells were treated with a series of concentrations of the compound and illuminated for 10 min (laser wavelength of 660 nm and laser power density of 40 mW/cm2). (B) Dark toxicity of Pc-1, Pc-2, Pc-3, Pc-4 and Pc-5 toward A431 cells without light illumination. | |
{kind=link}
To verify the phototoxicity of these conjugates visually, the morphological changes in A431 cells after PDT treatment with these conjugates were observed using microscopy. As shown in Fig. 6, without PDT treatment, the cells showed unabridged shapes, while the cells treated by incubation with conjugates, followed by irradiation for 10 min with a 660 nm-laser, showed significant morphological changes, including cellular swelling, bleb formation, and vesicle rupture. The PDT activities of these conjugates were distinct.

|
Download:
|
| Fig. 6. Cell morphological changes observed by microscopy before and after PDT for A431 cells treated with Pc-1, Pc-2, Pc-3, Pc-4 and Pc-5. | |
{kind=link}
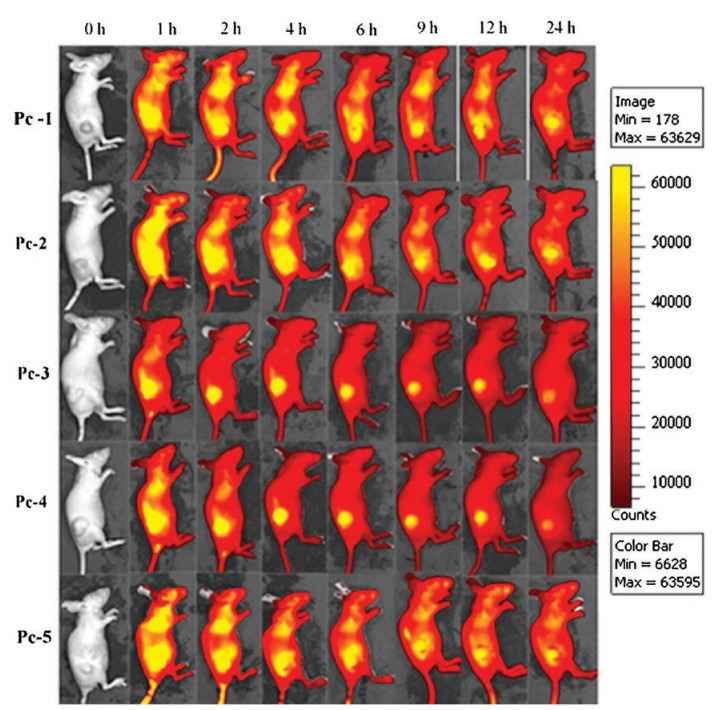
The in vivo distributions of the conjugates in tumor-bearing mice were monitored continuously with an in vivo fluorescence imaging system (IVIS Lumina Ⅱ, Xenogen, Alameda, CA, USA). The strong NIR fluorescence of the Pc component greatly facilitated the monitoring. As shown in Fig. 7, all Pcs injected into the mice were quickly distributed throughout the body approximately one hour post-injection. The signals accumulated in the tumor area quickly, while those in the normal tissues began to decrease rapidly. All Pcs showed good tumor accumulation, and enrichment consistently occurred within several hours of injection. Compared with other three Pcs, Pc-3 and Pc-5 showed relatively better tumor-targeting capabilities. The mice injected with Pc-3 or Pc-5 showed quick accumulation of the intense fluorescence signal in the tumor area and a rapid decrease in the background fluorescence signal in normal tissue. At only 2 h post-injection, the signals were located almost entirely in the tumor area; negligible signals were detected in other tissues, which resulted in excellent fluorescence imaging contrast that could clearly distinguish tumors from the surrounding normal tissue. The intense fluorescence signal in the tumor site could be maintained for more than 12 h post-injection. The mice injected with Pc-1 and Pc-2 also showed a very strong fluorescence signal in the tumor site but relatively stronger background fluorescence signals in normal tissues, particularly in the liver. This lower contrast may be due to the relatively higher hydrophobicity of the Pc component. The mice injected with Pc-4 showed much slower clearance of the background fluorescence signals than did the other mice. The relatively long hexaethylene glycol monomethyl ether substitution near the Pc component may afford the conjugate a longer plasma half-life, and longer times were required for the non-enriched conjugate to be filtered from the plasma. The in vivo distribution results may indicate that Pc-3 and Pc-5 have relatively better enrichment capabilities. Both of them can well accumulate in tumor sites with weak background intensity, and may have good potential for in vivo tumor imaging and tumor-targeted PDT.

|
Download:
|
| Fig. 7. In vivo imagings of Pc-1-5 in xenograft mice bearing human A431 tumors. BALB/C nude mice bearing A431 tumors were intravenously injected with 25 nmol/mouse of Pcs via the tail vein. Photographs were taken at 0, 1, 2, 4, 6, 9, 12, and 24 h after injection of Pcs. One representative mouse from each group (n = 4) is shown. | |
{kind=link}
Pc-3 showed relatively higher PDT activity and better tumor cell selectivity in the cell-based MTT assay than did the other conjugates. In addition, Pc-3 and Pc-5 showed better tumor enrichment in the in vivo distribution assay than did the three other conjugates. Thus, we selected Pc-3 as the photosensitizer for further evaluating the tumor treatment potential by in vivo PDT experiments. Human A431 tumors were first inoculated in the leg of BALB/c nude mice. When tumors reached 200 mm3, the mice were randomly divided into two groups (n = 5 per group), with each injected with 50 nmol/mouse of Pc-3 or PBS as a control, followed by irradiation with a 660-nm laser (200 mW for 15 min) 2 h after injection. Tumor growth and weight were measured for each mouse. As shown in Fig. 8A and B, the tumors in group 1 developed scabs, which gradually decreased in size and disappeared approximately 10 days after PDT. No tumors had regrown 40 days after the PDT treatment. One PDT treatment with Pc-3 appears to completely abolish tumors in the mouse xenograft tumor model. Compared with those in group 1, the tumors grew very rapidly in group 2, indicating that irradiation alone had no influence on the inhibition of tumor growth. Fig. 8C shows tumor-growth curves for mice receiving PDT treatment with Pc-3 or light irradiation only. Fig. 8D shows the weights of the mice receiving PDT treatment. The weight of the mice showed no obvious differences among the groups, indicating that irradiation with Pc-3 had no distinct side effect on mouse weight and may thus meet the necessary safety criteria for in vivo tumor treatment application.

|
Download:
|
| Fig. 8. In vivo photodynamic therapeutic efficacy on mice bearing A431 tumor xenografts: 50 nmol/mouse of Pc-3 or PBS was injected intravenously, and irradiation was performed at 150 mW/cm2 for 16 minutes (laser wavelength of 660 nm) two hours after injection (five mice in each group). All mice were treated with PDT only once. (A) Representative images of mice injected with Pc-3 treated by 660-nm illumination. (B) Representative images of mice injected with PBS treated by 660-nm illumination. (C) Tumor-growth curves of mice injected with Pc-3 and PBS. (D) Body weight of mice injected with Pc-3 and PBS on different days. Data shown are means ± SEM (n = 5). | |
{kind=link}
In conclusion, we developed EGFR receptor-specific photosensitizers based on peptide-conjugated highly water-soluble Pc conjugates. Here, we report the synthesis, photophysical property analysis, and in vitro and in vivo PDT studies of these Pc conjugates. Among the conjugates, Pc-3 possessed better properties for discriminating between healthy and tumor tissues and abolished inoculated tumors in the subcutaneous xenograft tumor model, making this approach a promising therapeutic agent for the treatment of cancer.
AcknowledgmentsThis research was supported by the National Key R&D Program of China (No. 2017YFC1104400), the Major Program of the National Natural Science Foundation of China (No. 31527801) and the National Natural Science Foundation of China (No. 81773293).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.04.025.
| [1] |
(a) P. Agostinis, K. Berg, K. A. Cengel, et al., CA-Cancer J. Clin. 61(2011) 250-281; (b) A. P. Castano, P. Mroz, M. R. Hamblin, Nat. Rev. Cancer 6(2006) 535-545. |
| [2] |
(a) J. P. Celli, B. Q. Spring, I. Rizvi, et al., Chem. Rev. 110(2010) 2795-2838; (b) M. R. Detty, S. L. Gibson, S. J. Wagner, J. Med. Chem. 47(2004) 3897-3915; (c) L. L. Rui, H. L. Cao, Y. D. Xue, et al., Chin. Chem. Lett. 27(2016) 1412-1420. |
| [3] |
(a) M. Ethirajan, Y. Chen, P. Joshin, R. K. Pandey, Chem. Soc. Rev. 40(2011) 340-362; (b) M. Mitsunaga, M. Ogawa, N. Kosaka, et al., Nat. Med. 17(2011) 1685-1691; (c) Z. Meng, B. Shan, L. Zhang, et al., Chin. Chem. Lett. 27(2016) 623-626. |
| [4] |
(a) Z. Jiang, J. Shao, T. Yang, J. Wang, L. Jia, J. Pharm. Biomed. 87(2014) 98-104; (b) L. B. Josefsen, R. W. Boyle, Theranostics 2(2012) 916-966. |
| [5] |
A.E. O'Connor, W.M. Gallagher, A.T. Byrne, Photochem. Photobiol. 85 (2009) 1053-1074. DOI:10.1111/php.2009.85.issue-5 |
| [6] |
N.B Mckeown, Phthalocyanine Materials:Synthesis, Structure and Function[M]. UK: Cambridge University Press, 1998.
|
| [7] |
(a) M. C. DeRosa, R. J. Crutchley, Chem. Rev. 233(2002) 351-371; (b) C. N. Lunardi, A. C. Tedesco, Curr. Org. Chem. 9(2005) 813-821. |
| [8] |
(a) F. Dumoulin, M. Durmus, V. Ahsen, T. Nyokong, Coord. Chem. Rev. 254(2010) 2792-2847; (b) S. Singh, A. Aggarwal, D. K. Bhupathiraju, et al., Chem. Rev. 115(2015) 10261-10306. |
| [9] |
(a) S. Makhseed, M. Machacek, W. Alfadly, et al., Chem. Commun. 49(2013) 11149-11151; (b) J. Alzeer, B. R. Vummidi, P. J. C. Roth, N. W. Luedtke, Angew. Chem. Int. Ed. 48(2009) 9362; (c) Y. Zorlu, M. A. Ermeydan, F. Dumoulin, et al., Photochem. Photobiol. Sci. 8(2009) 312-318; (d) H. Li, T. J. Jensen, F. R. Fronczek, M. G. H. Vicente, J. Med. Chem. 51(2008) 502-511; (e) W. Liu, T. J. Jensen, F. R. Fronczek, et al., J. Med. Chem. 48(2005) 1033-1041. |
| [10] |
A.M. Bugaj, Photochem. Photobiol. Sci. 10 (2011) 1097-1109. DOI:10.1039/c0pp00147c |
| [11] |
(a) L. Luan, W. Fang, W. Liu, et al., Org. Biomol. Chem. 14(2016) 2985-2992; (b) Z. Chen, P. Xu, J. Chen, et al., Acta Biomater. 10(2014) 4257-4268; (c) E. Ranyuk, N. Cauchon, K. Klarskov, B. Gue'rin, J. E. van Lier, J. Med. Chem. 56(2013) 1520-1534; (d) B. G. Ongarora, K. R. Fontenot, X. Hu, et al., J. Med. Chem. 55(2012) 3725-3738; (e) P. Xu, J. Chen, Z. Chen, et al., PloS One 7(2012) e3705. |
| [12] |
(a) R. Mahato, W. Tai, K. Cheng, Adv. Drug Deliv. Rev. 63(2011) 659-670; (b) A. Srivatsan, M. Ethirajan, S. K. Pandey, et al., Mol. Pharm. 8(2011) 1186-1197. |
| [13] |
(a) Y. Li, J. Wang, X. Zhang, et al., Org. Biomol. Chem. 13(2015) 7681-7694; (b) F. Li, Q. Liu, Z. Liang, et al., Org. Biomol. Chem. 14(2016) 3409-3422. |
| [14] |
(a) G. M. Fang, J. X. Wang, L. Liu, Angew. Chem. Int. Ed. 51(2012) 10347-10350; (b) M. Pan, S. Gao, Y. Zheng, et al., J. Am. Chem. Soc. 138(2016) 7429-7435. |
| [15] |
(a) Y. Guo, H. Yuan, W. L. Rice, et al., J. Am. Chem. Soc. 134(2012) 19338-19341; (b) F. Liu, D. Deng, X. Chen, et al., Mol. Imaging Biol. 12(2010) 595-607. |
| [16] |
(a) K. Viehweger, L. Barbaro, K. P. García, et al., Bioconjug. Chem. 25(2014) 1011-1022; (b) S. X. Song, D. Liu, J. L. Peng, et al., Int. J. Pharm. 363(2008) 155-161. |