 2018, Vol. 29
2018, Vol. 29 



b Comprehensive AIDS Research Center, School of Medicine, Tsinghua University, Beijing 100084, China
The acquired immunodeficiency syndrome (AIDS) is a worldwide infectious disease caused by human immunodeficiency virus (HIV) [1-3]. HIV-1, a type of HIV with high pathogenicity is the major causative agent for the AIDS pandemic [4]. The development of an effective vaccine against HIV-1 infection remains elusive [5, 6]. Thus anti-HIV drugs have played a crucial role in AIDS treatment. HIV-1 fusion inhibitors are important anti-HIV-1 drugs for AIDS therapy. They overcome the side effects and drug resistance caused by long-term use of reverse transcriptase inhibitors and protease inhibitors [7-9]. Gp41, a glycoprotein on the surface of HIV-1, is the target for HIV-1 fusion inhibitor [10]. When HIV-1 initiates the fusion process, gp41 tunes its conformation to hairpin structure, forming a stable 6-helix bundle (6HB) structure [11, 12]. This conservative conformation change can be blocked by HIV-1 fusion inhibitors [13]. Currently, enfuvirtide (T20) approved by the FDA in 2003 is the only HIV-1 peptide fusion drug with high activity, few side effects and clear mechanism of action [14, 15]. However, the widespread use of T20 in the clinical application has induced appearance of strong T20-resistance in patients [16, 17]. In addition, T20 as a peptide drug derived from the C-terminal heptad repeat region (CHR) of gp41, presents short half-life in vivo, limiting its usage in the treatment of HIV-1 infection [18].
To overcome these shortcomings, many new fusion inhibitors such as SFT, C34, T649v and SC34EK have be developed via structural optimization of the peptide derived from CHR region [19-21]. Among these reported fusion inhibitors, electrostatically constrained peptide SC34EK is one of the most active molecules (29-fold more active than T20) [22, 23]. Further study showed that SC34EK also presented enhanced activity against T20-resistant virus [24]. However, the helical structure of SC34EK is sensitive to proteases cleavage in vivo, which is a bottleneck problem that needs to be overcome. Herein, we reported novel SC34EK variants prepared by inserting hydrocarbon staples into SC34EK. Hydrocarbon staples were found to significantly improve the activity and protease stability of SC34EK, leading to a potent fusion inhibitor SC34EK-1 with IC50 value of 0.04–6.4 nmol/L against diverse HIV-1 subtypes. In addition, we explained the possible reason why SC34EK-1 was more active than SC34EK.
Inspired by Walensky's work to improve the protease stability of T649v via introducing two staples at the C- and N-terminus of the inhibitor [25], we adopted the stapling strategy to improve the properties of SC34EK. By incorporation of two α-methyl, α-alkenyl amino acids (S5) at i and i + 4 positions of peptide sequence [26], an all-hydrocarbon staple was formed in the peptide after adding the olefin metathesis catalyst to the resin-bound peptide. Stapled peptides have been successfully applied in the regulation of p53 pathway [27], NOTCH pathway [28], Wnt pathway [29] and HCV infection [30]. However, the effect of introducing staples into SC34EK on its activity and the stability has not been reported yet.Therefore, we designed novel SC34EK variants with a stable helical conformation stabilized by all-hydrocarbon staples.
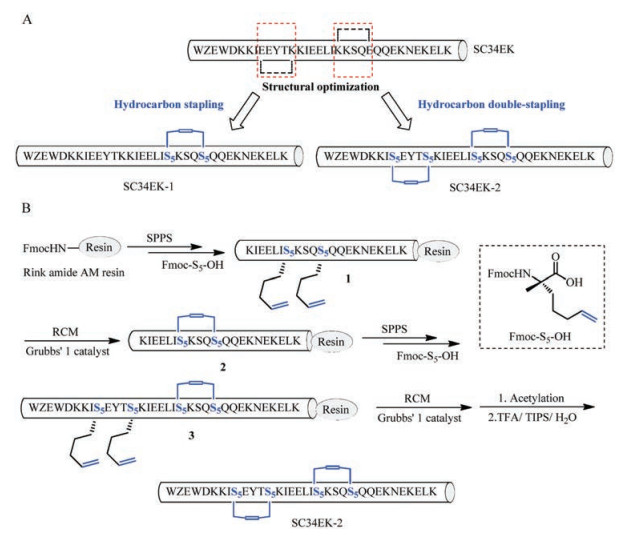
To choose the positions for stapling, we analyzed the critical amino acids involved in the interaction between SC34EK and gp41 [31]. We avoided these critical amino acids and replaced Lys647 and Glu651 used for forming the salt bridge in SC34EK using two α-methyl, α-alkenyl amino acids to generate SC34EK-1 with one staple. Furthermore, we replaced Glu636 and Lys640, Lys647 and Glu651 with α-methyl, α-alkenyl amino acids to obtain SC34EK-2 with two staples. The designed peptides were synthesized using the Fmoc (9H-fluorenyl-methoxycarbonyl) solid-phase peptide synthesis (Fig. 1) [32-35]. All synthetic peptides had a C-terminal amidation and an N-terminal acetylation. After identification by mass spectrometry and purification by HPLC, stapled SC34EKs with 95% purity were obtained.

|
Download:
|
| Fig. 1. (A) Structural optimization of SC34EK through hydrocarbon stapling strategy. Z represents norleucine, an artificial amino acid. Dotted lines indicate salt bridges existing in SC34EK. (B) Solid-phase synthetic route for preparing stapled SC34EK with two staples. | |
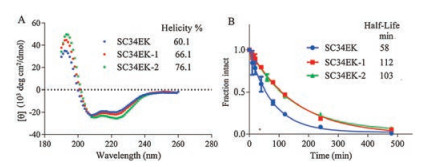
With SC34EK and its stapled peptides in hand, the impact of introducing staples on SC34EK peptide α-helicity was determined by CD. As shown in Fig. 2A, there was a positive absorption peak at 195 nm and two negative absorption peaks at 208, 222 nm of the CD spectrum, indicating that SC34EK adopted a stable helical structure in phosphate buffer [23]. The two stapled peptides showed the same characteristic absorption peaks in the CD spectrum, suggesting that they also formed stable helical structures. The relative percent helicity of SC34EK, SC34EK-1 and SC34EK-2 was calculated to be 60.1%, 66.1% and 76.1%, respectively, using the helicity equation [36]. These data implied that the introduction of staple in the SC34EK could increase its α-helicity, and two staples inserted in the 636, 640 and 647, 651 positions was more conducive to the α-helicity improvement. Above results supported Verdine's view that the staples inserted in peptides could promote the formation of the helical structure [37].

|
Download:
|
| Fig. 2. (A) CD spectra and a-helicity of SC34EK and stapled SC34EKs in PBS (pH 7.5). (B) Proteolytic stability of SC34EK and stapled SC34EKs against chymotrypsin, monitored by HPLC. Both CD and digestion assays were performed 3 times. | |
To explore the effect of staples on the stability of SC34EK against proteolytic enzymes, SC34EK and its stapled peptides (25 μmol/L) were exposed to 0.5 ng/μL chymotrypsin at 30 ℃. The degradation of peptide was monitored by HPLC. Degradation curves of SC34EK and its stapled peptides were displayed in Fig. 2B. The half-life of SC34EK was calculated to be 58 min. Nevertheless, the half-life of T649v, another fusion inhibitor containing 35 amino acids was only 14 min at the same digestion condition, reflecting 4.1-fold decrease in chymotrypsin resistance compared to SC34EK. This may be due to a significant distinction for α-helicity between SC34EK and T649v. The α-helicity of SC34EK was 60.1%, while the value for T649v was 13%. Because proteases require the substrate adopt an extended conformation to hydrolyze the amide bond, a stable alpha-helical structure can bring substrate protease resistance. Thus SC34EK had a stronger enzyme resistance than T649. Singly stapled SC34EK-1 exhibited the longest half-life of 112 min, indicating a 1.9-fold enhancement in stability against chymotrypsin compared to SC34EK, while doubly stapled SC34EK-2 displayed a longer half-life of 103 min, surpassing SC34EK by 1.8-fold. The similar chymotrypsin resistance of SC34EK-1 and SC34EK-2 was probably due to their similar helicity. The increased half-lives of stapled peptides suggested that the introduction of staples could enhance the protease stability of SC34EK. This observation was also consistent with previous study that staples could enhance the stability of peptides against protease [25].
To confirm whether the stapled SC34EK-1 and SC34EK-2 can effectively inhibit HIV-1 infection like their parent peptide SC34EK, we evaluated the activity of SC34EK and its stapled peptides against HIV-1 using different subtypes of HIV-1 pseudoviruses standard strains and epidemic strains in China. For HIV-1 epidemic strains, CNE6 and CNE11 belong to B' subtype, CNE15 and CNE30 belong to BC subtype, CNE5 and CNE55 belong to CRF01_AE subtype. For HIV-1 standard strains, sf162, JRFL and HXB2 all are B subtype. Ghost(3)X4/R5 cells were infected with nine HIV-1 pseudoviruses containing luciferase reporter gene, in the presence of peptide inhibitors respectively. The ability of HIV-1 pseudoviruses to infect cells was determined by measuring the luciferase reporter activity.
The half maximal inhibitory concentration (IC50) of SC34EK and its stapled peptide against HIV-1infecting cells was presented in Table 1. SC34EK possessed nanomolar-level inhibitory activity against six endemic strains and three standard strains, consistent with previous report that it was a potent HIV-1 fusion inhibitor. By comparing IC50 values of SC34EK and its stapled peptides, we found that SC34EK-1 and SC34EK-2 not only had nanomolar-level inhibitory activity against all HIV-1 tested, but also showed 1.1– 52.5 fold improved activity than SC34EK. These results indicated that the introduction of staples into SC34EK could significantly increase its anti-HIV-1 activity. Further analysis of the effect of stapling position and number of staples inserting in SC34EK on the anti-HIV-1 activity, it was revealed that the anti-HIV-1 activity of singly stapled SC34EK-1 and doubly stapled SC34EK-2 was respectively 2.5–52.5 fold and 1.1–7.0 fold higher than that of SC34EK. This data showed that the introduction of a staple at 647, 651 position played more critical impact on the anti-HIV-1 activity of SC34EK than the introduction of two staples at 636, 640 and 647, 651 position. SC34EK-1 with IC50 value of 0.04–6.4 nmol/L against nine HIV-1 pseudoviruses, was the most potent fusion inhibitor among the tested peptides in this study. Surprisingly, introduction of one staple in the SC34EK dramatically improved its antiviral activity.
|
|
Table 1 Inhibitory activity of SC34EK and stapled SC34EKs against HIV-1 variants. |

To examine whether SC34EK-1 could inhibit T20-resistant HIV-1 variants, T20-resistant HIV-1NL-4-3 pseudoviruses carrying single mutation or double mutations were constructed. We tested the inhibitory activity of T20, SC34EK and SC34EK-1 against these resistant HIV-1 variants respectively, and the activity data was listed in Table 2. In contrast to the inhibitory activity of T20 against the tested HIV-1NL-4-3 pseudoviruses and its variants, we found that both SC34EK and SC34EK-1 were able to efficiently inhibit T20-resistant strains. For the D36G mutant strain, it significantly enhanced the HIV-1 pseudovirion sensitivity to T20, leading to a 30-fold enhancement in the inhibitory activity of T20. However, the inhibitory activities of SC34EK and SC34EK-1 towards D36G mutants were lower than natural strains, indicating that these stapled peptides were insensitive to D36G mutants. The IC50 values of SC34EK for the six single mutations D36G, I37T, V38A, V38M, Q40H, N43K T20 resistant strains ranged from 0.8–2.7 nmol/L, whereas IC50 values of SC34EK-1 for these mutant strains were 0.7–2.6 nmol/L. This data insinuated that singly stapled SC34EK-1 retained the ability of SC34EK to efficiently inhibit single mutation T-20 resistant strains. For three double mutations I37T/N43K, G36S/V38M and V38A/N42T, the IC50 values of SC34EK and SC34EK-1 were measured to be 1.2–44.3 nmol/L, and 2.1–17.2 nmol/L, suggesting that SC34EK-1 could inhibit the two mutation T-20-resistant strains as efficiently as SC34EK.
|
|
Table 2 Inhibitory activity of SC34EK and stapled SC34EKs against T20-resistant HIV-1 variants. |

{kind=link}
{kind=link}
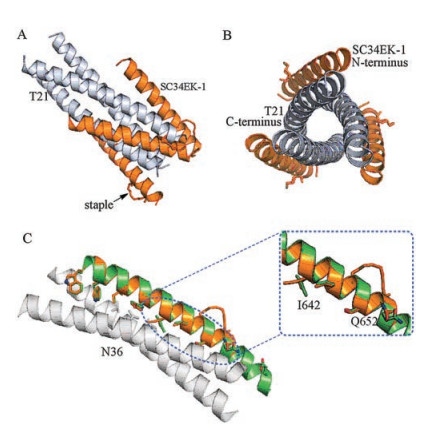
To understand why SC34EK-1 was more active than SC34EK, we prepared the crystals of SC34EK-1 and T21 complex, via utilizing the siting-drop vapor-diffusion method at 18 ℃. The crystal structure of the complex with a resolution of 1.8 Å was determined by using the molecular replacement method and taking the HIV gp41 core crystal structure (PDB 1ENV) [38] as a search model. View from front or side of the SC34EK-1/T21 complex, SC34EK-1 and T21 formed a typical 6-HB structure in which the N-terminus of SC34EK-1 interacted with the C-terminus of T21 (Figs. 3A and B). These observed details of the SC34EK-1/T21 complex were as same as the reported 6-HB structure of SC34EK/ N36 complex (N36 derived from the CHR domain, similar to T21). As anticipated, a hydrocarbon staple between 647, 651 position was found in the solvent exposed surfaces of SC34EK-1, indicating the location of this staple is consistent with our design. Next, structure alignment of SC34EK-1 with SC34EK gave the structural changes generated by the introduction of staple in SC34EK. It was regrettable that structure of SC34EK-1 and SC34EK could not be completely aligned, due to lack of 655-661 of SC34EK-1. The backbone structure showed that SC34EK-1 presented the same helical manner as SC34EK (Fig. 3C). The RMSD value of SC34EK-1 and SC34EK over 27 Cα atom was 0.327 Å, also suggesting that SC34EK-1 and SC34EK possessing same backbone structure. Further comparing the conformations of amino acids on the interaction surface between SC34EK-1/SC34EK and N36, we found that the conformations of Trp628, Trp631, Ile635, Tyr638, Leu645 and Ser649 in SC34EK-1/SC34EK were the same. However, there was an observable difference in the side chain conformations of Ile642 and Gln652 (Fig. 3C). The side-chain conformations of Ile642 and Gln652 in SC34EK-1 were closer to N36 than that of SC34EK, implying that the interaction between SC34EK-1 and N36 was stronger. Insertion of single staple might cause the conformational changes that promote the interaction of SC34EK and its target, which would explain the enhanced anti-HIV-1 activity of SC34EK-1.

|
Download:
|
| Fig. 3. Crystal structure of SC34EK-1 complex with T21. (A) Crystal structure of SC34EK-1 (orange) and T21 (light purple, refer to NHR) viewed from the side. Orange sticks indicate staple observed in the crystal structure. (B) Crystal structure of SC34EK-1 (orange) and T21 (gray) viewed from the N-terminus of SC34EK-1. (C) Crystal structure of SC34EK (green, PDB code: 2Z2T) bound to N36 (gray, refer to NHR) and comparison with SC34EK-1 (orange). Interacted residues were shown as stick models. | |
{kind=link}
To summarize, we developed singly and doubly stapled SC34EK, namely, SC34EK-1 and SC34EK-2 that possessed higher helicity than the native peptide. Chymotrypsin digest assays showed that the introduction of staples in SC34EK could prolong its half-life to 103–112 min. More importantly, the stapled SC34EKs could exhibit more effectively inhibit infection of the diverse HIV-1 variants, compared to SC34EK. SC34EK-1 was the most potent fusion inhibitor with a lower IC50 value of 0.04–6.4 nmol/L against the HIV-1 epidemic strains and standard strains in antivirus assay. The following anti-T20 resistant study demonstrated that SC34EK-1 retained high potency against T20-resistant strains. The determination of SC34EK-1/T21 complex structure confirmed that insertion of single staple at 647, 651 position could alter the configurations of the amino acids on the interacting surface, thus improving the anti-HIV-1 activity of SC34EK-1. Our work on stapled HIV fusion inhibitors would provide not only a potential candidate SC34EK-1 with high anti-HIV-1 activity and strong enzyme stability for HIV-1 treatment, but also a practical strategy for the future design and synthesis of potent inhibitors against others viruses such as respiratory syncytial virus (RSV) and acute respiratory syndrome (SARS).
AcknowledgmentsWe thank Professor Yuxian He for the help on T20-resistance test and Professor Xinquan Wang for SC34EK-1/T21 complex structure determination. We thank Dr. Linqi Zhang for his kind support and helpful suggestion. This work was supported by the National Natural Science Foundation of China (No. 21602121), the Natural Science Foundation of Inner Mongolia (No. 2016BS0201), the Inner Mongolia Autonomous Region Higher School Youth scientific Talents Support Project(No. NJYT-17-B22), the Research Funds of Baotou Medical College (Nos. BSJJ201620, BYJJ-YF 201707) and Beijing Tongzhou District Science and Technology Project (No. KJ2017CX039-14).
| [1] |
R.J. Pomerantz, D.L. Horn, Nat. Med. 9 (2003) 867-873. DOI:10.1038/nm0703-867 |
| [2] |
S.G. Deeks, S.R. Lewin, D.V. Havlir, Lancet 382 (2013) 1525-1533. DOI:10.1016/S0140-6736(13)61809-7 |
| [3] |
G. Maartens, C. Celum, S.R. Lewin, Lancet 384 (2014) 258-271. DOI:10.1016/S0140-6736(14)60164-1 |
| [4] |
P.M. Sharp, B.H Hahn, Cold Spring Harb. Perspect. Med 1 (2011) a006841. |
| [5] |
T. Bradley, J. Pollara, S. Santra, et al., Nat. Commun (2017), 15711. |
| [6] |
L. Leal, C. Lucero, J.M. Gatell, et al., Expert. Rev. Vaccines. 16 (2017) 587-600. DOI:10.1080/14760584.2017.1322513 |
| [7] |
J.J. Tan, X.T. Ma, C. Liu, Curr. Pharm. Des. 19 (2013) 1810-1817. DOI:10.2174/1381612811319100005 |
| [8] |
M. Fumakia, S. Yang, J. Gu, et al., Rev. Med. Virol. 26 (2016) 4-20. DOI:10.1002/rmv.1853 |
| [9] |
D. Zhang, W. Li, S.B. Jiang, Expert. Opin. Ther. Pat. 25 (2015) 159-173. DOI:10.1517/13543776.2014.987752 |
| [10] |
C.G. Pan, S.W. Liu, S.B. Jiang, J. Formos. Med. Assoc. 109 (2010) 94-105. DOI:10.1016/S0929-6646(10)60029-0 |
| [11] |
M. Lu, S.C. Blacklow, P.S. Kim, Nat. Struct. Biol. 2 (1995) 1075-1082. DOI:10.1038/nsb1295-1075 |
| [12] |
Y.X. He, Curr. Pharm. Des. 19 (2013) 1800-1809. DOI:10.2174/1381612811319100004 |
| [13] |
F. Yu, L. Lu, L. Du, et al., Viruses 5 (2013) 127-149. DOI:10.3390/v5010127 |
| [14] |
J.P. Lalezari, K. Henry, O'Hearn M., et al., N. Engl. J. Med. 348 (2003) 2175-2185. DOI:10.1056/NEJMoa035026 |
| [15] |
J.M. Kilby, S. Hopkins, T.M. Venetta, Nat. Med. 4 (1998) 1302-1307. DOI:10.1038/3293 |
| [16] |
M.D. Miller, D.J. Hazuda, Drug Resist. Updat. 7 (2004) 89-95. DOI:10.1016/j.drup.2004.03.003 |
| [17] |
M.L. Greenberg, N. Cammack, J. Antimicrob. Chemother. 54 (2004) 333-340. DOI:10.1093/jac/dkh330 |
| [18] |
L. Cai, C. Pan, L. Xu, et al., FASEB J. 26 (2012) 1018-1026. DOI:10.1096/fj.11-195289 |
| [19] |
Y.X. He, Y.H. Xiao, H.F. Song, et al., J. Biol. Chem. 283 (2008) 11126-11134. DOI:10.1074/jbc.M800200200 |
| [20] |
D.C. Chan, C.T. Chutkowski, P.S. Kim, Proc. Natl. Acad. Sci. U. S. A. 95 (1998) 15613-15617. DOI:10.1073/pnas.95.26.15613 |
| [21] |
L.T. Rimsky, D.C. Shugars, T.J. Matthews, J. Virol. 72 (1998) 986-993. |
| [22] |
F. Miyamoto, E.N. Kodama, Antivir. Chem. Chemother. 22 (2012) 151-158. DOI:10.3851/IMP1930 |
| [23] |
A. Otaka, M. Nakamura, D. Nameki, et al., Angew. Chem. Int. Ed. 41 (2002) 2937-2940. DOI:10.1002/1521-3773(20020816)41:16<2937::AID-ANIE2937>3.0.CO;2-J |
| [24] |
K. Shimura, D. Nameki, K. Kajiwara, J. Biol. Chem. 285 (2010) 39471-39480. DOI:10.1074/jbc.M110.145789 |
| [25] |
G.H. Bird, N. Madani, A.F. Perry, et al., Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 14093-14098. DOI:10.1073/pnas.1002713107 |
| [26] |
C.E. Schafmeister, J. Po, G.L. Verdine, J. Am. Chem. Soc. 122 (2000) 5891-5892. DOI:10.1021/ja000563a |
| [27] |
F. Bernal, A.F. Tyler, S.J. Korsmeyer, et al., J. Am. Chem. Soc. 129 (2007) 2456-2457. DOI:10.1021/ja0693587 |
| [28] |
R.E. Moellering, M. Cornejo, T.N. Davis, et al., Nature 462 (2009) 182-188. DOI:10.1038/nature08543 |
| [29] |
(a) T. N. Grossmanna, J. T. H. Yeha, B. R. Bowman, et al., Proc. Natl. Acad. Sci. U. S. A. 109(2012) 17942-17947; (b) H. K. Cui, B. Zhao, Y. H. Li, et al., Cell Res. 23(2013) 581-584. |
| [30] |
H.K. Cui, J. Qing, Y. Guo, et al., Bioorg. Med. Chem. 21 (2013) 3547-3554. DOI:10.1016/j.bmc.2013.02.011 |
| [31] |
H. Nishikawa, S. Nakamura, E. Kodama, et al., Int. J. Biochem. Cell Biol. 41 (2009) 891-899. DOI:10.1016/j.biocel.2008.08.039 |
| [32] |
Y.W. Kim, T.N. Grossmann, G.L. Verdine, Nat. Protoc. 6 (2011) 761-771. DOI:10.1038/nprot.2011.324 |
| [33] |
G.M. Fang, Y.M. Li, F. Shen, et al., Angew. Chem. Int. Ed. 50 (2011) 7645-7649. DOI:10.1002/anie.201100996 |
| [34] |
G.M. Fang, J.X. Wang, L. Liu, Angew. Chem. Int. Ed. 51 (2012) 10347-10350. DOI:10.1002/anie.201203843 |
| [35] |
J.S. Zheng, S. Tang, Y.K. Qi, et al., Nat. Protoc. 8 (2013) 2483-2495. DOI:10.1038/nprot.2013.152 |
| [36] |
D. Wang, K. Chen, Kulp Ⅲ J.L., P.S. Arora, J. Am. Chem. Soc. 128 (2006) 9248-9256. DOI:10.1021/ja062710w |
| [37] |
G.L. Verdine, G.J Hilinski, Drug Discov. Today Technol 9 (2012) e1-e70. |
| [38] |
W. Weissenhorn, A. Dessen, S.C. Harrison, et al., Nature 387 (1997) 426-430. DOI:10.1038/387426a0 |