 2018, Vol. 29
2018, Vol. 29 


 ,
Rui Wangb
,
Rui Wangb
2 Laboratory of Preclinical Study for New Drugs of Gansu Province, School of Basic Medical Sciences, Lanzhou University, Lanzhou 730000, China
Antimicrobial peptides, which are widely distributed among microorganisms, animals and plants, form an evolutionarily conserved component of an organism's innate immune system. Despite great diversity in their primary sequences, antimicrobial peptides share certain established hallmarks, including cationicity, hydrophobicity and amphiphilicity [1, 2]. Among antimicrobial peptides, helical peptides are the most widely distributed and possess various pharmacological properties, such as antimicrobial, antiviral and anticancer activities [1, 2]. It is generally believed that the main mechanism of action of antimicrobial peptides is membrane disruption via a series of physical processes; as a result, there is a relatively low likelihood that cells will develop greater resistance to these peptides [1, 3, 4].
Although antimicrobial peptides are attractive therapeutics due to their particular mechanisms of action, their applications are limited by obvious obstacles, such as toxicology profiles, potency and proteolytic degradation [5, 6]. To overcome these problems, extensive efforts have been dedicated to obtaining ideal antimicrobial peptides via modification, the incorporation of unnatural amino acids, peptidomimetics and multimerization [6, 7]. Dimerization has been shown to be an effective strategy for producing antimicrobial peptides with greater therapeutic efficacy than their monomeric equivalents [8]. In prior studies, homo- and heteromeric antimicrobial peptides have been synthesized by connecting two identical or different monomeric peptides using a linker, and such dimeric peptides have exhibited significantly enhanced antimicrobial activity [9-14].
Anoplin, which was isolated from Anoplius samariensis venom, is considered the smallest natural α-helical antimicrobial peptide with potent activity against both Gram-positive and Gramnegative bacteria [15]. Mastoparan, which was isolated from Vespula lewisii venom, is an α-helical antimicrobial peptide with a wide variety of biological effects [16, 17]. Anoplin and mastoparan have been extensively used as templates in structure-activity relationship studies. TP10, which is a cell-penetrating peptide with a remarkable capacity for membrane translocation, is constructed by linking a 6-residue segment from the neuropeptide galanin with mastoparan via an additional Lys residue [18, 19]. In our prior studies, a series of analogs of TP10 were designed to explore the effects of structural changes on these peptides' cell-penetrating and antimicrobial activities [20, 21]. In the present work, we designed and synthesized a group of heteromeric antimicrobial peptides by utilizing different amino acids, including leucine, proline and aminocaproic acid, to connect anoplin and mastoparan (Table 1). Compared with leucine, proline and aminocaproic acid are potent α-helix breakers and can increase the flexibility of peptides. The main objective of this work was to investigate the effects of the flexibility conferred by proline and aminocaproic acid on the potency and selectivity of these heteromeric antimicrobial peptides.
|
|
Table 1 Sequences and physical features of peptides. |
Interestingly, a number of studies have shown that the introduction of a proline residue close to the center of a sequence can contribute to dissociating the antimicrobial and hemolytic activities of α-helical antimicrobial peptides, despite the fact that this alteration breaks the helical structure to varying extents [22-27]. In the present study, the introduction of proline or aminocaproic acid as the linker amino acid led to significant decreases in the α-helical contents of AM-2 and AM-3 compared with AM-1 (Fig. S1 in Supporting information). However, the anticancer and antimicrobial activities of AM-2 and AM-3 were significantly higher than those of AM-1, for which the linker amino acid was leucine (Table 2). Unfortunately, all tested heteromeric antimicrobial peptides exhibited a marked increase in hemolytic activity compared with anoplin and mastoparan (Table 2). This phenomenon may have been caused by the heteromeric peptides' increased length, hydrophobicity and helicity, which have been proven to be correlated with increasing hemolytic activity for α-helical antimicrobial peptides [22, 28, 29]. Although the results of the hemolysis assay were unsatisfactory, due to the use of proline and aminocaproic acid as the linker amino acid, the hemolytic activities of AM-2 and AM-3 were significantly lower than that of AM-1. In addition, we used a series of assays to explore the mechanisms of action of these heteromeric peptides. As shown in Figs. S2 and S3 in Supporting information, all the heteromeric antimicrobial peptides could kill cancer cells and bacterial cells in a short time, suggesting that similarly to most naturally occurring antimicrobial peptides, the tested peptides induced membrane damage.
|
|
Table 2 Anticancer activity, antimicrobial activity and hemolytic activity of peptides. |
Why did the flexibility conferred by the linker amino acids increase the potency and selectivity of the heteromeric antimicrobial peptides AM-2 and AM-3 compared with AM-1? Although certain prior studies have confirmed that greater flexibility could increase the potency and selectivity of antimicrobial peptides [24, 27], more effort will be required to provide a mechanistic explanation for this phenomenon. However, it is difficult to observe interactions between antimicrobial peptides and membranes at the molecular level using traditional experimental methods. Therefore, we used molecular dynamics (MD) simulations to explore and compare the effects of different linker amino acids on interactions of the aforementioned heteromeric antimicrobial peptides with different membranes.
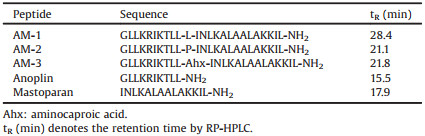
Because the erythrocyte membrane is mainly composed of zwitterionic phospholipids, hydrophobic interactions are the major driving force for the binding of antimicrobial peptides to this membrane [30, 31]. Generally, beyond an optimal hydrophobicity window, higher hydrophobicity is correlated with stronger hemolytic activity [31, 32]. Measuring the retention time on RPHPLC is an effective approach that can reflect the actual hydrophobic properties of peptides better than evaluations of mean hydrophobicity [33]. As shown in Table 1, the hydrophobicity of AM-2 and AM-3 was significantly lower than that of AM-1. Why did the introduction of proline or aminocaproic acid result in a decrease in hydrophobicity for AM-2 and AM-3? As shown in Fig. 1, simulations of peptides in water demonstrated that AM-2 and AM-3 adopted a compact hairpin conformation in which hydrophobic residues were partially shielded from the water. However, the structure of AM-1 was more extensive than those of AM-2 and AM-3. Therefore, it became difficult for the hydrophobic residues of AM-2 and AM-3 to interact with the reversed-phase matrices (the C18 layer). Consequently, AM-2 and AM-3 were eluted from the RP-HPLC column earlier than AM-1. The decreased hydrophobicity of AM-2 and AM-3 implied that the forces driving the binding of these peptides to the erythrocyte membrane were weaker than those of AM-1, resulting in decreased hemolytic activity (Table 2).

|
Download:
|
| Fig. 1. Snapshots presenting the changes of configurations of AM-1-AM-3 in water. The main chains of peptide are represented by cartoon, colored by gray. The side chains (stick) of hydrophilic residues are yellow, those of hydrophobic residues are orange and those of cationic residues are blue. | |
{kind=link}
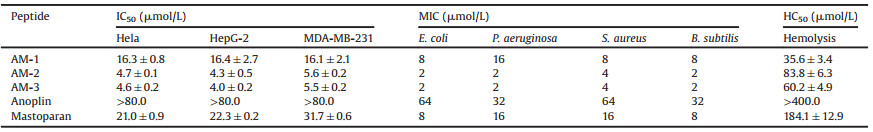
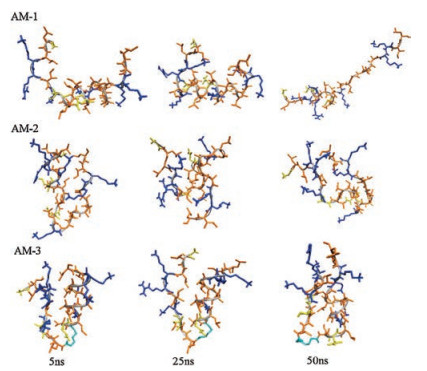
To date, relatively few studies have addressed the effects of helical breakers on the interactions of antimicrobial peptides with membranes after adsorption. Therefore, we used MD simulations to provide additional details about how linker amino acids affected these interactions for the examined heteromeric antimicrobial peptides. First, we investigated interactions of these heteromeric antimicrobial peptides with a zwitterionic POPC membrane that mimics the erythrocyte membrane. As shown in Fig. S4A in Supporting information, significantly faster insertion into the POPC lipid bilayer was observed for AM-1 than for AM-2 and AM-3; this phenomenon may have occurred because AM-1 exhibited higher hydrophobicity. After entering the hydrophobic region of the lipid bilayer, AM-1 maintained a fully helical structure, whereas AM-2 and AM-3 had bent structures induced by proline and aminocaproic acid, respectively (Fig. 2A). Therefore, the adoption of a more helical structure may have contributed to greater interaction with the POPC membrane for AM-1 than for AM-2 and AM-3. The degree of membrane thinning and disordering can be used to reflect the extent of interaction of antimicrobial peptides with membranes [34, 35]. As shown in Fig. 2B and Fig. S4B in Supporting information, upon insertion into the POPC lipid bilayer, AM-1 induced more membrane thinning and disordering than AM-2 and AM-3. Therefore, the flexible structure conferred by the use of proline or aminocaproic acid as the linker amino acid could decrease interactions of AM-2 and AM-3 with the zwitterionic POPC lipid bilayer. Overall, the increased flexibility of AM-2 and AM-3 due to the presence of a helical breaker could result in low hemolytic activity by decreasing these peptides' ability to bind to erythrocyte membranes and their interactions with these membranes upon insertion.

|
Download:
|
| Fig. 2. (A) Snapshots presenting the final conformations of AM-1–AM-3 in 200 ns simulations of peptides in the POPC lipid bilayer. The bilayers are showed by lines, colored with gray (POPC), the large balls (CPK) represent the phosphorus atoms of the lipid head groups. The side chains (stick) of hydrophilic residues are yellow, those of hydrophobic residues are orange and those of cationic residues are blue. (B) Mass densities and average thickness of membrane. The thickness of membrane was defined as the average bilayer interface distance. The densities of lipids, peptide and water are colored by black, red and green, respectively. | |
{kind=link}
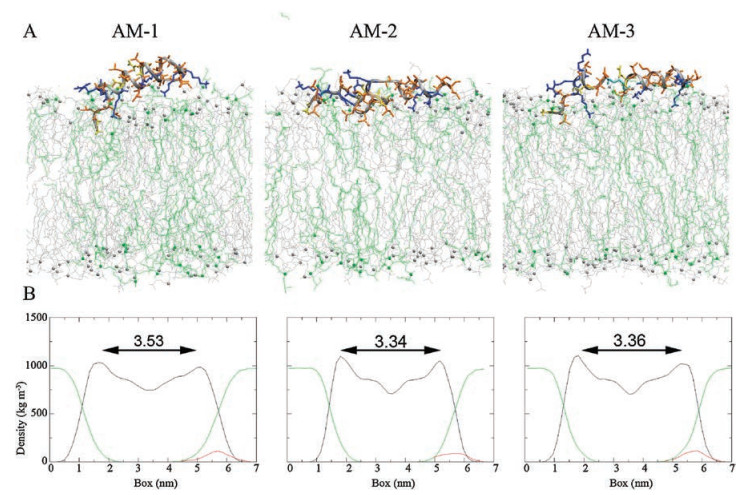
In addition, we investigated interactions of these heteromeric antimicrobial peptides with a mixed POPG/POPC lipid bilayer that mimics the membranes of bacterial cells or cancer cells, which are rich in acidic phospholipids [36]. As shown in Fig. 3A and Fig. S5A in Supporting information, none of the heteromeric peptides fully entered the mixed lipid bilayer, likely due to the presence of negatively charged POPG and to running simulations for overly short times. Nevertheless, the MD simulations showed that upon adsorption, AM-2 and AM-3 induced more membrane thinning and disordering effects than AM-1 (Fig. 3B and Fig. S5B in Supporting information), indicating that the former peptides exhibited stronger interactions with mixed membranes. This phenomenon may have occurred because due to their flexible structures, AM-2 and AM-3 had more hydrophobic residues that could interact with the POPG/POPC lipid bilayer than AM-1 (Fig. 3A). Accordingly, these results indicate that AM-2 and AM-3 exhibited more significant cytotoxicity to cancer cells and bacterial cells than AM-1.

|
Download:
|
| Fig. 3. (A) Snapshots presenting the final conformations of AM-1–AM-3 in 200 ns simulations of peptides in the POPG/POPC lipid bilayer. The bilayers are showed by lines, colored with gray (POPC) and green (POPG). The side chains (stick) of hydrophilic residues are yellow, those of hydrophobic residues are orange and those of cationic residues are blue. (B) Mass densities and average thickness of membrane. | |
{kind=link}
Briefly, as linker amino acids, proline and aminocaproic acid can increase the flexibility of heteromeric antimicrobial peptides. This greater flexibility can cause heteromeric antimicrobial peptides to fold into a compact conformation in water, which decreases their hydrophobicity and their binding to erythrocyte membranes. In addition, our simulation results demonstrate that a flexible structure rather than a fully helical structure decreases interactions of antimicrobial peptides with zwitterionic membranes while increasing damage to negatively charged membranes. Consequently, a flexible structure can improve the potency of antimicrobial peptides for bacterial cells and cancer cells while reducing lytic activity with respect to red blood cells. In summary, these findings provide additional details about the effects of flexibility conferred by linker amino acids on the potency and selectivity of heteromeric antimicrobial peptides and will contribute to the design of efficient antimicrobial peptides.
AcknowledgmentsWe are grateful for the grants from the National Natural Science Foundation of China (Nos. 81773566, 21602092, 81473095), the Fundamental Research Funds for the Central Universities (Nos. lzujbky-2017-134, lzujbky-2017-120, lzujbky-2016-21). We also thank the Gansu Supercomputing Center of Cold and Arid Environment and Engineering Research Institute of Chinese Academy of Sciences for providing computational resource.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.04.011.
| [1] |
M.R. Yeaman, N.Y. Yount, Pharmacol. Rev. 55 (2003) 27-55. DOI:10.1124/pr.55.1.2 |
| [2] |
L.T. Nguyen, E.F. Haney, H.J. Vogel, Trends Biotechnol. 29 (2011) 464-472. DOI:10.1016/j.tibtech.2011.05.001 |
| [3] |
M. Zasloff, Nature 415 (2002) 389-395. DOI:10.1038/415389a |
| [4] |
G. Gabernet, A.T. Muller, J.A. Hiss, et al., MedChemComm 7 (2016) 2232-2245. DOI:10.1039/C6MD00376A |
| [5] |
C.D. Fjell, J.A. Hiss, R.E. Hancock, et al., Nat. Rev. Drug Discovery 11 (2011) 37-51. |
| [6] |
Z.Y. Ong, N. Wiradharma, Y.Y. Yang, Adv. Drug Delivery Rev. 78 (2014) 28-45. DOI:10.1016/j.addr.2014.10.013 |
| [7] |
N.K. Brogden, K.A. Brogden, Int. J. Antimicrob. Agents 38 (2011) 217-225. |
| [8] |
S.P. Liu, L. Zhou, R. Lakshminarayanan, R.W. Beuerman, Int. J. Pept. Res. Ther. 16 (2010) 199-213. DOI:10.1007/s10989-010-9230-z |
| [9] |
C.E. Dempsey, S. Ueno, M.B. Avison, Biochem. 42 (2003) 402-409. DOI:10.1021/bi026328h |
| [10] |
S.T. Yang, J.I. Kim, S.Y. Shin, Biotechnol. Lett. 31 (2009) 233-237. DOI:10.1007/s10529-008-9848-5 |
| [11] |
E.N. Lorenzon, G.F. Cespedes, E.F. Vicente, et al., Antimicrob. Agents Chemother. 56 (2012) 3004-3010. DOI:10.1128/AAC.06262-11 |
| [12] |
M. Nishida, Y. Imura, M. Yamamoto, et al., Biochemical 46 (2007) 14284-14290. DOI:10.1021/bi701850m |
| [13] |
J. Zerweck, E. Strandberg, J. Burck, et al., Eur. Biophys. J. Biophy. 45 (2016) 535-547. DOI:10.1007/s00249-016-1120-7 |
| [14] |
W.Y. Li, N.M. O'Brien-Simpson, J. Tailhades, et al., Chem. Biol. 22 (2015) 1250-1258. DOI:10.1016/j.chembiol.2015.08.011 |
| [15] |
K. Konno, M. Hisada, R. Fontana, et al., BBA-Protein Struct. M 1550 (2001) 70-80. DOI:10.1016/S0167-4838(01)00271-0 |
| [16] |
Y. Hirai, Y. Ueno, T. Yasuhara, et al., Biomed. Res.-Tokyo 1 (1980) 185-187. DOI:10.2220/biomedres.1.185 |
| [17] |
M. Moreno, E. Giralt, Toxins 7 (2015) 1126-1150. DOI:10.3390/toxins7041126 |
| [18] |
U. Soomets, M. Lindgren, X. Gallet, et al., BBA-Biomembr. 1467 (2000) 165-176. DOI:10.1016/S0005-2736(00)00216-9 |
| [19] |
S. El-Andaloussi, P. Jarver, H.J. Johansson, et al., Biochem. J. 407 (2007) 285-292. DOI:10.1042/BJ20070507 |
| [20] |
J.J. Song, M. Kai, W. Zhang, et al., Peptides 32 (2011) 1934-1941. DOI:10.1016/j.peptides.2011.07.018 |
| [21] |
J.Q. Xie, Y.M. Gou, Q. Zhao, et al., J. Pept. Sci. 21 (2015) 599-607. DOI:10.1002/psc.v21.7 |
| [22] |
Y.X. Chen, C.T. Mant, S.W. Farmer, et al., J. Biol. Chem. 280 (2005) 12316-12329. DOI:10.1074/jbc.M413406200 |
| [23] |
Y.M. Song, Y. Park, S.S. Lim, et al., Biochem.-US 44 (2005) 12094-12106. DOI:10.1021/bi050765p |
| [24] |
L.S. Vermeer, Y. Lan, V. Abbate, et al., J. Biol. Chem. 287 (2012) 34120-34133. DOI:10.1074/jbc.M112.359067 |
| [25] |
S.T. Yang, J.Y. Lee, H.J. Kim, et al., FEBS J. 273 (2006) 4040-4054. DOI:10.1111/j.1742-4658.2006.05407.x |
| [26] |
L.J. Zhang, R. Benz, R.E.W. Hancock, Biochemistry 38 (1999) 8102-8111. DOI:10.1021/bi9904104 |
| [27] |
S. Bobone, G. Bocchinfuso, Y. Park, et al., J. Pept. Sci. 19 (2013) 758-769. DOI:10.1002/psc.v19.12 |
| [28] |
A. Hollmanna, M. Martinez, M.E. Noguera, et al., Colloid Surf. B 141 (2016) 528-536. DOI:10.1016/j.colsurfb.2016.02.003 |
| [29] |
L.H. Kondejewski, M. Jelokhani-Niaraki, S.W. Farmer, et al., J. Biol. Chem. 274 (1999) 13181-13192. DOI:10.1074/jbc.274.19.13181 |
| [30] |
K. Matsuzaki, BBA-Biomembr. 1788 (2009) 1687-1692. DOI:10.1016/j.bbamem.2008.09.013 |
| [31] |
Y.X. Chen, M.T. Guarnieri, A.I. Vasil, et al., Antimicrob. Agents Chemother. 51 (2007) 1398-1406. DOI:10.1128/AAC.00925-06 |
| [32] |
Y.B. Huang, J.F. Huang, Y.X. Chen, Protein Cell 1 (2010) 143-152. DOI:10.1007/s13238-010-0004-3 |
| [33] |
S. Kim, S.S. Kim, B.J. Lee, Peptides 26 (2005) 2050-2056. DOI:10.1016/j.peptides.2005.04.007 |
| [34] |
C.W. Tsai, N.Y. Hsu, C.H. Wang, et al., J. Mol. Biol. 392 (2009) 837-854. DOI:10.1016/j.jmb.2009.06.071 |
| [35] |
J.J. Song, W. Zhang, M. Kai, et al., Mol. Pharm. 10 (2013) 2934-2941. DOI:10.1021/mp400052s |
| [36] |
D.W. Hoskin, A. Ramamoorthy, BBA-Biomembr. 1778 (2008) 357-375. DOI:10.1016/j.bbamem.2007.11.008 |