 2018, Vol. 29
2018, Vol. 29 



b School of Life Science and Engineering, Southwest Jiaotong University, Chengdu 611756, China;
c Department of Gynecology, The Second Clinical Medical College of Jinan University, Shenzhen People's Hospital, Shenzhen 518020, China;
d Key Lab in Healthy Science and Technology, Division of Life Science, Shenzhen Graduate School of Tsinghua University, Shenzhen 518055, China
Overexpression of estrogen receptors (ER-α and ER-β, nuclear receptor superfamily members) is observed in over 70% of all breast cancer cases [1]. Upon the stimulation of estradiol, ER-α regulates many genes to prompt cell proliferation and metastasis [2]. Selective estrogen receptor modulators (SERMs) designed as competitive inhibitors to target estradiol binding pocket, such as tamoxifen and raloxifen, are broadly used as efficient ER positive breast cancer therapeutics [3]. However, in addition to increasing drug resistance, SERMs suffer from severe adverse side effects, including higher uterine cancer and stroke risks [4]. Therefore, targeting non-ligand binding sites of ER-α is viewed as a promising strategy to overcome the drawbacks of SERMs.
The nuclear receptor-cofactor interactions are generally based on the consensus a-helical motifs of cofactors, which bind to a hydrophobic cleft on AF-2 surface of nuclear receptors [5]. Several helical peptides containing the LXXLL (L: leucine and X: any amino acid) have been developed as inhibitors of ER-coactivator interactions [6]. However, cellular activities of these peptides have not been well reported [7].
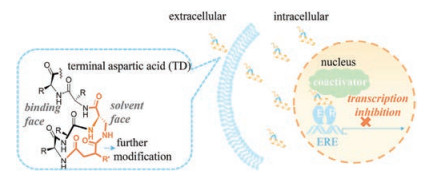
Recently, we developed a facile N-terminus helix-nucleating strategy using an unnaturally tethered aspartic acid (TD strategy). A free amino group is intentionally preserved on the tethered aspartic acid for further modification, which exposes to solvent face by designing (Fig. 1). This modification was similar to those accomplished at the backbone positions [8].

|
Download:
|
| Fig. 1. Terminal aspartic acid stabilized peptide conjugate perturbs the interaction of estrogen receptor α and coactivator, thus inhibits the transcription of estrogen receptor α and induces cell death. | |
{kind=link}
Peptidomimetic estrogen receptor modulators (PERMs) constructed using this strategy show improved therapeutic properties [9]. However, relatively weak nuclear translocation efficiency of TD PERM limits its further biological applications.
Herein, we optimized TD peptides by attachment of an arginine rich cell-penetrating peptide (Ac-RRWWRRWRR-NH2) WR onto the preserved on-tether free amino group [10]. The conjugated peptide showed improved cell permeability and nuclear translocation efficiency, and subsequently presented selective cellgrowth inhibition to ER-α-positive cell lines.
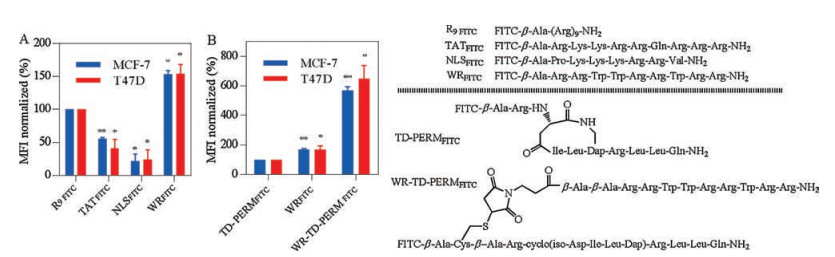
All of the peptides were synthesized by Fmoc strategy and conjugated peptides were obtained via Michael addition chemistry (Schemes S1 and S2 in Supporting information). High isoelectric point counts for peptides' efficient nucleus translocation. Accordingly, Arg-rich cell penetrating peptides R9FITC [11], TATFITC [12], NLSFITC [13] and WRFITC [10] (FITC-labelled peptides were named followed by a FITC) were tested for their cellular uptakes in ER-α positive cell lines MCF-7 and T47D (Fig. 2). According to fluorescence activated cell sorting (FACS) results, WRFITC showed better cellular uptake than TATFITC and R9FITC. Then WR was attached to TD PERM peptide previously reported to generate WR-TD-PERMFITC as shown in Fig. 2A. Quantification of the cellular uptake indicated that WR-TD-PERMFITC showed over 6-fold cellular uptake increment than TD-PERMFITC itself in both cell lines (Fig. 2B). Based on fluorescence polarization assays, WR-TD-PERMFITC showed comparable binding affinity to PERM, indicating the WR attachment did not disturb the binding of the peptide to ER-α, as it was designed to point at the solvent face (Fig. S1 in Supporting information).

|
Download:
|
| Fig. 2. (A) Cellular uptake of FITC labelled cell-penetrating peptides in both MCF-7 and T47D cell lines. Cells were treated by 5 μmol/L peptides for 1 h, then washed by PBS for 3 times. 105 cells were collected for each analysis. The mean fluorescent intensity (MFI) of each analysis was calculated and normalized to R9FITC peptide. (B) Cellular uptakes of peptide conjugates in both MCF-7 and T47D cell lines. Cells were treated by 2.5 μmol/L peptides for 1 h, then washed by PBS for 3 times. 105 cells were collected for each analysis. The mean fluorescent intensity (MFI) of each analysis was calculated and normalized to TD-PERMFITC peptide. Error bars represent the standard error of mean from more than two independent experiments. *P < 0.05, **P < 0.01. | |
{kind=link}
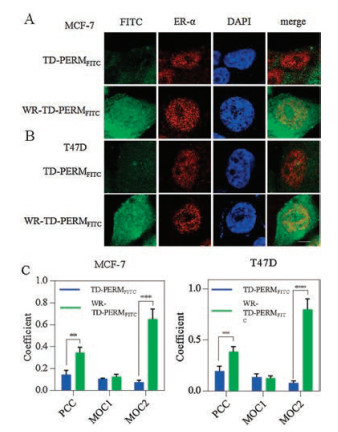
We then assessed the cellular distribution of TD-PERMFITC and WR-TD-PERMFITC using confocal laser scanning microscopy (CLSM). Significantly improved nucleus accumulation was observed for WR-TD-PERMFITC in both MCF-7 and T47D cell lines (Figs. 3A and B, respectively). WR-TD-PERMFITC showed increased co-localization with ER-α in the nucleus according to the Pearson correlation coefficient (PCC) and Manders overlap coefficient (MOC) [14] (Fig. 3C). The correlation between incidence of the peptide and the target (PCC), and the ratio of the target overlapped by the peptide (MOC2) of the conjugated peptide (WR-TDPERMFITC) was higher compared to the unmodified peptide (TD-PERMFITC). The change suggested stronger interactions between the peptide and the target. The attachment of WR on the preserved on-tether free amino group significantly improved PERM's cellular uptake and target association.

|
Download:
|
| Fig. 3. (A) Cellular distribution of WR-TD-PERM peptide. Confocal imaging of TD-PERMFITC and WR-TD-PERMFITC in (A) MCF-7 and (B) T47D cell lines. The nucleus was stained by DAPI. The cellular ER-α was first immunized by anti-ER-α primary antibody and was then immunized by Texas red-conjugated goat anti-rabbit IgG antibody. (C) Co-localization analysis between peptides and ER-α. Coefficient found per peptide for more than 5 individual cells, from more than two independent experiments. **P < 0.01, ***P < 0.005. | |
{kind=link}
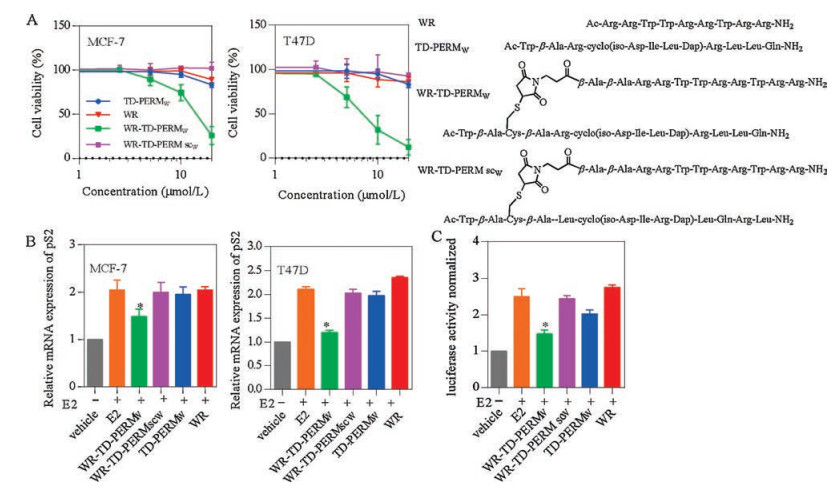
WR-TD-PERMW (Trp-labelled peptides were named with a following W for concentration determination [15]) was then prepared for cellular activity studies. MCF-7 and T47D are the common oestrogen receptor a positive cell lines and here we used them for cellular activity studies. WR-TD-PERMW showed satisfying growth inhibition activities of LD50 at ~12 μmol/L and 7 μmol/L for MCF-7 and T47D cells lines after 24 h treatment. While the unconjugated sequence TD-PERMW, WR or scrambled sequence WR-TD-PERMscW (sc represent scrambled) show minimal activity as shown in Fig. 4A. According to the LDH release results (Fig. S2 in Supporting information) and toxicity results of ER-negative cells (MDA-MB-231) and normal cells (HEK293T) (Fig. S3 in Supporting information), WR-TD-PERMW showed low toxicity at effective concentrations for ER-positive cell lines.

|
Download:
|
| Fig. 4. Cellular activity assays. (A) Growth inhibition of MCF-7 and T47D cell lines after 24 h treatment of peptides. (B) Inhibition of transcriptional activities of ER-α pS2 mRNA level upon treatment with peptides in ER-positive cells (MCF-7 and T47D). Cells were cultured with 2.5 μmol/L of the indicated peptides in the presence of 100 nmol/L estradiol (E2) (unless indicated). Cells were lysed after 20 h of estradiol treatment, after which RNA was isolated and prepared for RT-qPCR analysis of endogenous ER-mediated pS2 mRNA levels (normalized). (C) HEK293T cells were transfected with pEGFP-C1-ER-α plasmid (addgene) with PGMER-Lu ER reporter plasmid (Qcbio) which contained the ERE motif. Luciferase activities are expressed as the ratio of Firefly luciferase signal to Renilla luciferase signal and are normalized to the ratio obtained when cells were solely treated with the vehicle. Error bars represent the standard error of mean from more than two independent experiments. *P < 0.05. | |
{kind=link}
ER-α transcriptional activity was then tested upon peptide treatment, transcriptional activities of ER-α with peptide treatment at non-toxic concentrations (2.5 μmol/L) were tested with luciferase assays and quantitative polymerase-chain reaction (RT-qPCR) (Figs. 4C and D). The results revealed that WR-TDPERMW efficiently down-regulated the ER-α-mediated transcription, while unconjugated peptide TD-PERMW, WR or scrambled peptide WR-TD-PERMscW showed negligible effects on pS2 gene expression under 2.5 μmol/L concentration.
This proof of concept study clearly indicated the uptakepromoting modification on the preserved amino group could significantly enhance the TD-PERM's cell membrane permeability co-localization with target, growth inhibition and transcriptional activity. This study verified the effectiveness of TD-PERM in modification way and the good modification ability of preserved amino group.
In summary, utilizing the intentionally preserved on-tether free amino group in the N-cap template, a cell penetrating peptide was attached to TD-PERM peptide without interrupting its target binding ability. The resulting peptide conjugation showed significantly improved cellular uptake and target association, which resulted in satisfying biological activity towards ER-α positive breast cancer cells. This proof-of-concept study validates the merit of preserved on-tether amino group and directs the further development of TD-PERM.
AcknowledgmentsWe acknowledge financial support from the National Natural Science Foundation of China (Nos. 21778009 and 81701818; MOST2015DFA31590); the Shenzhen Science and Technology Innovation Committee (Nos. JCYJ20170412150719814 and GJHS20170310093122365).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.04.004.
| [1] |
B. Fisher, J.P. Costantino, D.L. Wickerham, et al., J. Natl. Cancer Inst. 97 (2005) 1652-1662. DOI:10.1093/jnci/dji372 |
| [2] |
(a) T. Nasomyon, S. Samphao, S. Sangkhathat, et al., Anticancer Res. 34 (2014) 1333-1342; (b) S. J. Howell, S. R. Johnston, A. Howell, Best Pract. Res. Clin. Endocrinol. Metab. 18 (2004) 47-66; (c) A. D'Assoro, R. Busby, I. Acu, et al., Oncogene 27 (2008) 3901-3911; (d) R. L. Dillon, R. Marcotte, B. T. Hennessy, et al., Cancer Res. 69 (2009) 5057-5064. |
| [3] |
(a) B. Fisher, J. P. Costantino, D. L. Wickerham, et al., J. Natl. Cancer Inst. 90 (1998) 1371-1388; (b) Y. J. Kwon, D. R. Hurst, A. D. Steg, et al., Clin. Exp. Metastasis 28 (2011) 437-449. |
| [4] |
(a) L. Bernstein, D. Deapen, J. R. Cerhan, et al., J. Natl. Cancer Inst. 91 (1999) 1654-1662; (b) Y. Shang, M. Brown, Science 295 (2002) 2465-2468. |
| [5] |
A.K. Shiau, D. Barstad, P.M. Loria, et al., Cell 95 (1998) 927-937. DOI:10.1016/S0092-8674(00)81717-1 |
| [6] |
(a) J. M. Hall, C. Y. Chang, D. P. McDonnell, Mol. Endocrinol. 14 (2000) 2010-2023; (b) A. M. Leduc, J. O. Trent, J. L. Wittliff, et al., Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 11273-11278; (c) A. K. Galande, K. S. Bramlett, J. O. Trent, et al., ChemBioChem 6 (2005) 1991-1998; (d) T. Phan, H. D. Nguyen, H. Göksel, et al., Chem. Commun. 46 (2010) 8207-8209; (e) C. Phillips, L. R. Roberts, M. Schade, et al., J. Am. Chem. Soc. 133 (2011) 9696-9699. |
| [7] |
(a) M. Carraz, W. Zwart, T. Phan, et al., Chem. Biol. 16 (2009) 702-711; (b) K. Tints, M. Prink, T. Neuman, et al., Int. J. Mol. Sci. 15 (2014) 5680-5698. |
| [8] |
J.S. Zheng, M. Yu, Y.K. Qi, et al., J. Am. Chem. Soc. 136 (2014) 3695-3704. DOI:10.1021/ja500222u |
| [9] |
(a) H. Zhao, Q. S. Liu, H. Geng, et al., Angew. Chem. Int. Ed. 128 (2016) 12267-12272; (b) Y. H. Jiang, Q. W. Deng, H. Zhao, et al., ACS Chem. Biol. 13 (2018) 628-635; (c) M. S. Xie, H. Zhao, Q. S. Liu, et al., J. Med. Chem. 60 (2017) 8731-8740. |
| [10] |
D. Delaroche, B. Aussedat, S. Aubry, et al., Anal. Chem. 79 (2007) 1932-1938. DOI:10.1021/ac061108l |
| [11] |
N.A. Alhakamy, P. Dhar, C.J. Berkland, Mol. Pharm. 13 (2016) 1047-1057. |
| [12] |
M. Silhol, M. Tyagi, M. Giacca, et al., Eur. J. Biochem. 269 (2002) 494-501. DOI:10.1046/j.0014-2956.2001.02671.x |
| [13] |
M.A. Zanta, P. Belguise-Valladier, J.P. Behr, Proc. Natl. Acad. Sci. U. S. A. 96 (1999) 91-96. DOI:10.1073/pnas.96.1.91 |
| [14] |
S. Bolte, F. Cordelieres, J. Microsc. 224 (2006) 213-232. DOI:10.1111/jmi.2006.224.issue-3 |
| [15] |
Y. Kim, T.N. Grossmann, G.L. Verdine, Nat. Protoc. 6 (2011) 761-771. DOI:10.1038/nprot.2011.324 |