 2018, Vol. 29
2018, Vol. 29 



RAGE is a 43 kDa type Ⅰ TM receptor that belongs to the immunoglobulin superfamily [1], which is expressed in a wide range of tissues, including brain, liver, and heart, where it activates pro-inflammatory signaling as a part of the innate immune system in response to external stress [2]. Most previous work focuses on determining the structural basis for dimerization of RAGE, which includes the V-domain that is responsible for binding AGE, as well as the V-C1-C2 fragment that is responsible for binding the S100A-family ligands [3, 4]. A recent study using a yeast two-hybrid approach suggests that the RAGE cytoplasmic domain interacts with diaphanous-1 (Dia1), and this binding partner is found to constitute the basis for intracellular signaling [5]. Pin-Chuan Su and Bryan W. Berger use AraC-based Transcriptional Reporter Assay (AraTM) to identify the key JM interactions, in which they demonstrated that the intracellular region of RAGE has a great influence on the dimerization activity of RAGE [6]. The TM-JM domain is increasingly becoming a vital factor in determining structure and oligomerization of RAGE. However, much less is known about specific TM-JM interactions occurring in the oligomeric states of RAGE. There are a number of experimental approaches to investigate the assembling intensity and transformation of the TMD dimerization, including TOXCAT assays [7, 8], co-immunoprecipitation, and FRET methods [9]. Nowadays, molecular dynamic (MD) simulation has been popularly used to explore the structure and dynamics with extreme detail, literally on the atomistic level. The simulation technology is innovated rapidly to adapt a definite biology system, such as the Martini force field [10, 11], which is excelsior in simulating the long scale assembling process and evaluating the interaction between the proteins near or embedded in the phospholipid bilayers [12-14].
In this study, we explored the underlying interaction mechanism of RAGE TM domains by multiscale simulation methods. We mainly used a coarse-grained model to simulate the dimer conformation and investigated the role of JM-A375 in determining the RAGE TMD structure. We show the full length and various segments of RAGE and reveal the sequence of TM, CYTO and JMA375 included in the CYTO. While the JM domain with the JM-A375 was added on the TM domains, a stable TM dimeric conformation was remained. The effect of the JM domains attributes to the stabilization of the anionic lipids in the inner leaflet, which shows the lipid-mediated mechanism on the dimerization and bioactivities of RAGE.
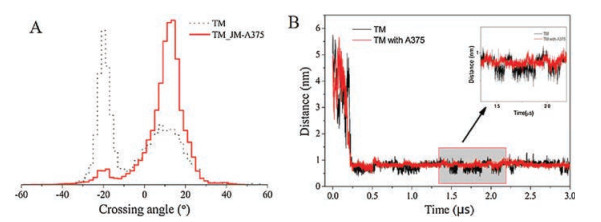
We first examined the dimerization status of TMs and the important residues affecting dimerization. The crossing angle of the twomonomers wasmonitored and calculated (Fig. 1A) to investigate the structural features of the wild-type TMs of the RAGE receptor during the assembling process. RAGE dimers were found to exhibit the inter-converted dimeric configurations, with the peaks of the crossing angle distribution of the RAGE located at about -22° and 12°. The results implied that the dimer transformed from the righthanded mode to the parallel mode alternatively.

|
Download:
|
| Fig. 1. Two dimeric conformations of the RAGE-TM domains. (A) The dimers show two main crossing angles, corresponding to their respective contact interfaces. The residue contact matrixes of the predominant negative-angle dimers and the positive-angle dimers. (B) The evolvements of the crossing angle and the distance between the monomers as a function of simulation time. (C) A helical wheel representation of TM sequence: left-handed form, which was packed by the six main residues of A342xxxG346xxG349xxxT353xxL356xxxV360 (filled by green); right-handed form, which was packed by the four main residues of L345xxxG349xxG352xxxL356 (filled by purple). | |
{kind=link}
When observing the crossing angle and distance as a function of time (Fig. 1B), we found that the crossing angle and distance have the same trend: When the angle was negative, the distance was between 0.6–0.7 nm, on the contrary, as the angle was positive, the distance was between 0.8–0.9 nm. Furthermore, the residue contacting distributions of the two alternative conformations were explored. The interacting residues involved in the righthanded conformation were detected to locate on the L345, G349, G352 and L356 positions (L345xxxG349xxG352xxxL356 motif) (Figs. 1A and C), while for the parallel conformation, the contacting residues appeared to balanced distribute and scatter along the entire transmembrane domain. Specifically, the residues of A342, G346, G349, T353, L356 and V360, notably occupied on the interface of the dimer and were well in line with the proposed heptad repeat motif A342xxxG346xxG349xxxT353xxL356xxxV360 (Figs. 1A and C).
After obtaining the residue interface, in order to obtain a better understanding of whether the complicated regions or residues play a substantial role in the two dimeric modes, mutations were conducted by substituting the residues by the Ile residues. Compared to wild type in which the crossing angle exhibited a bimodal distribution in which the peaks were located at about -22° and 12°, the crossing angle of G346IT353IV360I shows only a main peak sited at about -22° (Fig. S1A-left in Supporting information). In addition, the residue contact distribution of the mutant is a bit similar (left of Fig. 1A) which is corresponded with the right-handed configuration. Meanwhile, the mutation of L345IG352IG359I resulted in the disappear of the right-handed conformation (Fig. S1B in Supporting information). Therefore, the results proved our conjecture and indicated that the involved residues revealed by the simulations actually play an important role in the two modes, respectively.
Based on previous experiments, to evaluate the effect of JMA375 on RAGE dimerization, we extended the TM region to the region of the JM-A375 (364QRRQRRGEERKA375) (Fig. S2A in Supporting information). It is noteworthy that the structure of CT-RAGE has been reported in the literature [15], in which the structure of W363 to P376 (including JM-A375) is α-turn.
The interaction between the two peptides of TM_JM-A375 predominantly generated a solely left-handed dimeric configuration with a crossing angle of 12° (Fig. 2A). The residue contact distribution clearly showed the same membrane-spanning interaction residues in the TM left-handed configuration, and it is marked that some residues in the proximal membrane interact strongly (Fig. S2B in Supporting information). From the comparison chart of TM and TM-A375 distance along with time (Fig. 2B), we can intuitively see that TM-A375 is more stable in the simulation. As predicted, based on the above analysis, the most remarkable result to emerge was that TM-A375 is more stable in the simulation. The simulation results are in consistency with the existing evidence that the JM-A375 peptides improve the stability of dimeric assembly [15].

|
Download:
|
| Fig. 2. (A) The respective distance evolvements between the TM domains and TM-JM over the simulation time. (B) The left-handed conformation became the dominant conformation of the TM-JM dimer. | |
{kind=link}
The molecular dynamics simulations is feasible for us to explore the underlying mechanism why JM-A375 could stabilize the homo-dimerization of RAGE in molecular details [16]. Due to the fact that POPS lipid carries a net negative charge, R and K respectively carries a positive charge in JM-A375, we speculated that effect of the JM-A375 results from the electrostatic interaction between POPS and the R and K. As expected, the POPS tightly surrounded the peptide assembly (Fig. 3A). In contrast, the enrichment of POPC around the peptide assembly is not apparent (Fig. 3B). The different distribution of POPS and POPC therefore indicates the stronger interaction between POPS and JM-A375. To further resolute this interaction, we analyzed the frequency of contact between POPS and residues of JM-A375. It is obviously that the residues of R and K with positive charge predominantly interact with POPS, whereas the residues with negative charge and non-charged residues exhibit weak interaction with POPS (Fig. 3C). To better show the interaction of POPS and JM-A375, we used the VMD [17] to show the interaction details. During the simulation, the JM-A375 always showed a stronger contact with the POPS. In contrast, POPC is evenly distributed in the box with no specific interaction with the dimer (Fig. 3D).

|
Download:
|
| Fig. 3. The molecular interaction between POPS and JM-A375. (A, B) Number density of POPS and POPC around protein dimer. (C) The frequency of contact between the individual residue of JM-A375 and POPS. (D) Intuitive illustration of the interaction of POPS with JM-A375. | |
{kind=link}
We changed the membrane composition to run the simulations to further verify that the importance of the charged species of POPS in stabilizing the dimer conformation by interaction with the JM-A375. Keeping the system at an ionic neutrally station, the POPC/POPS mixed membrane was replaced with a pure POPC membrane (electroneutral membrane). As a result, the crossing angle of the dimeric conformation of RAGETM-JM (Fig. S3A in Supporting information) still showed two switching configurations, and the change of angle shows very unstable (Fig. S4B in Supporting information). Therefore, the influence of the JM-A375 was invalid if POPS lipid was removed. The results confirm that the electrostatic interactions play a decisive role in stabilizing the conformation of the RAGE_A375 dimer, and at the same time it can be concluded a difference of the membrane compositions have a significant effect on the interaction of the TM proteins.
The methods we used in this study are mainly coarse grained simulations. For details, please see Support information.
In summary, we explored the underlying interaction mechanism of RAGE TM domains by multiscale simulation methods. We mainly used a coarse-grained model to simulate the dimer conformation, revealing two switch packing patterns. With a series of site-directed mutagenesis at key interface sites, we further emphasized the key role of the A342xxxG346xxG349xxxT353xxL356xxxV360 motif in the left-hand configuration and the L345xxxG349xxG352xxxL356 motif in the right-hand configuration. In addition, we also investigated the role of JM-A375 in determining the RAGE TMD structure. When the JM domainwith the JM-A375 was added on the TM domains, a stable left-handed TM dimeric conformation was remained. The effect of the JM domains attributes to the stabilization of the anionic lipids in the inner leaflet, which shows the lipid-mediated mechanism on the dimerization and bioactivities of RAGE.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21672019, 21372026, 21402006) and the Fundamental Research Funds for the Central Universities (No. XK1701). This work was partly supported by CHEMCLOUDCOMPUTING.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.04.005.
| [1] |
A.M. Schmidt, S.D. Yan, S.F. Yan, D.M. Stern, J. Clin. Invest. 108 (2001) 949-955. DOI:10.1172/JCI200114002 |
| [2] |
M.A. Hofmann, S. Drury, C. Fu, et al., Cell 97 (1999) 889-901. DOI:10.1016/S0092-8674(00)80801-6 |
| [3] |
J. Xie, D.S. Burz, W. He, et al., J. Biol. Chem. 282 (2007) 4218-4231. |
| [4] |
H. Zong, A. Madden, M. Ward, et al., J. Biol. Chem. 285 (2010) 23137-23146. DOI:10.1074/jbc.M110.133827 |
| [5] |
B.I. Hudson, A.Z. Kalea, M. Del Mar Arriero, et al., J. Biol. Chem. 283 (2008) 34457-34468. DOI:10.1074/jbc.M801465200 |
| [6] |
P.C. Su, B.W. Berger, J. Biol. Chem. 287 (2012) 31515-31526. DOI:10.1074/jbc.M112.396895 |
| [7] |
C.R. Armstrong, A. Senes, Biochim. Biophys. Acta 1858 (2016) 2573-2583. DOI:10.1016/j.bbamem.2016.07.008 |
| [8] |
M. Chai, B. Liu, F. Sun, et al., Proteins 85 (2017) 1362-1370. DOI:10.1002/prot.v85.7 |
| [9] |
T.O. Peulen, O. Opanasyuk, C.A.M. Seidel, J. Phys. Chem. B 121 (2017) 8211-8241. DOI:10.1021/acs.jpcb.7b03441 |
| [10] |
J. Michalowsky, L.V. Schafer, C. Holm, J Smiatek, J. Chem. Phys. 146 (2017) 054501. DOI:10.1063/1.4974833 |
| [11] |
J.J. Uusitalo, H.I. Ingolfsson, S.J. Marrink, I. Faustino, Biophys. J. 113 (2017) 246-256. DOI:10.1016/j.bpj.2017.05.043 |
| [12] |
Z. Kong, H. Wang, L. Liang, et al., J. Mol. Mode 23 (2017) 113. DOI:10.1007/s00894-017-3292-1 |
| [13] |
F. Sun, L. Chen, P. Wei, Chai, et al., J. Chem. Inf. Model 57 (2017) 1375-1387. DOI:10.1021/acs.jcim.7b00196 |
| [14] |
F.D. Sun, X.F. Ding, L.D. Xu, et al., J. Phys. Chem. C 121 (2017) 17263-17275. DOI:10.1021/acs.jpcc.7b04347 |
| [15] |
P.C. Su, B.W. Berger, J. Mol. Biol. 425 (2013) 4652-4658. DOI:10.1016/j.jmb.2013.07.022 |
| [16] |
L. Wong, J. Bioinf. Comput. Biol 10 (2012) 1203002. DOI:10.1142/S0219720012030023 |
| [17] |
W. Humphrey, A. Dalke, K. Schulten, J. Mol. Graph. 14 (1996) 33-38. DOI:10.1016/0263-7855(96)00018-5 |