 2018, Vol. 29
2018, Vol. 29 



b Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China;
c Chonqqing Huapont Pharm. Co., Ltd., Chongqing 401121, China
Cyclic peptides from natural source are particularly important, they show an increasingly significant role in medicine and biology [1, 2], for example, vancomycin [3-5], cyclosporine [6], daptomycin [7] and teixobactin [8, 9]. Nowadays, hundreds of cyclic peptides are under clinic development, these peptides are used to treat various diseases, including bacillosis, tumor and autoimmune diseases [10].
Cyclic peptides are more stable than linear peptides with a variety of medical function and potential value. Among them, marine cyclopeptide is a kind of important lead compounds. Reniochalistatins A–E were first isolated from marine sponge Reniochalina stalagmitis, which was collected around Yongxing island in South China Sea by Lin's team in 2014 [11], and their structures were characterized by NMR and MALDI-TOF/TOF data. They found that the synthetic reniochalistatin E exhibited cytotoxic activity against myeloma RPMI-8226 and gastric MGC-803 cells with IC50 values of 4.9 μmol/L and 9.7 μmol/L, respectively.
Reniochalistatin E has attracted the interest from organic chemists since its isolation. The first total synthesis of reniochalistatin E was reported by Rafferty using liquid-phase peptide synthesis in 2017 with an overall 5.0% yield [12].
Here we describe a convergent synthesis of reniochalistatin E and its conformational isomers that utilized solid-phase peptide synthesis [13-19]. Reniochalistatin E is an octacyclopeptide containing three trans Pro moieties and a high percentage of hydrophobic residues, including two L-Iles, one L-Leu, one L-Val and one L-Trp. The three proline residues would control the conformation of cyclopeptide and affect its biological activities. For example, such effect was previously found with peptide sanguinamide B, which contains two proline residues and resulted in three configurations [20].
A retrosynthetic analysis of 1 is outlined in Scheme 1. Retrosynthetic planning starts with disconnecting the macrocycle at the Pro-Leu junction, so that a pro residue at the C-terminus would avoid epimerization during macrolactamization. The precursor would be linear peptide NH2-Leu-Trp(Boc)-Pro-ValIle-Pro-Ile-Pro-COOH 2a or NH2-Leu-Trp-Pro-Val-Ile-Pro-Ile-ProCOOH 2b, respectively.

|
Download:
|
| Scheme 1. Retrosynthetic analysis of reniochalistatin E (1). | |
The linear precursors 2a and 2b were synthesized on solid phase using 2-chlorotrityl chloride resin as the solid support [21]. As the macrocyclization step was the synthetic challenge of cyclopeptide synthesis, two cyclization precursor 2a and 2b were evaluated with or without the side chain protecting group. Both peptides 2a and 2b have a Pro residue at the C-terminus, which would minimize epimerization during cyclization. The two approaches were carefully examined.
The first amino acid was loaded by using N, N-diisopropyl ethylamine (DIPEA) as the base in anhydrous DMF overnight at room temperature. The resin was quenched by reacting with MeOH/DIPEA (10:1) for 1 h at room temperature. The Fmocprotected amino acids were activated by 2-(7-azabenzotriazol-1- yl)-N, N, N', N'-tetramethyluronium hexaflu-orophosphate (HATU)/DIPEA in DMF and reacted with Fmoc-deprotected resin bound peptide for 30 min or 1 h on a shaker. Before each coupling step, Fmoc deprotection of the resin bound peptide was achieved using 20% piperidine in DMF (Scheme 2).

|
Download:
|
| Scheme 2. The process of SPPS to synthesize cyclic peptide reniochalistatin E. | |
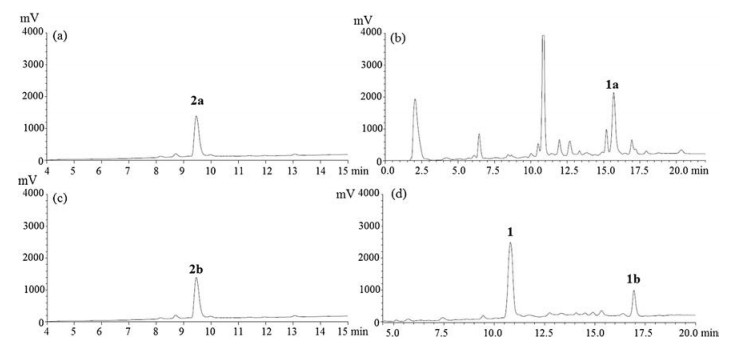
The linear peptide 3 was cleaved from resin by AcOH/TFE/DCM (4:1:1) or TFA/TIPS/H2O (95:2.5:2.5), respectively, affording peptides 2a and 2b, which were used without further purification. The TFA in solution was blown off by a stream of argon in a fume hood. Precooled diethyl ether was added to the mixture to precipitate the crude peptides. The suspension was centrifuged at 4500 r/min for 1 min at room temperature, and the pellet was collected and dried under vacuum. The linear peptide precursor 2a was obtained as pink powers with 92% yield (Fig. 1a), linear peptide 2b as white powers was obtained with 93% yield (Fig. 1c).

|
Download:
|
| Fig. 1. LC—MS analysis of the linear reniochalistatin E precursors and their cyclizations; UV was recorded at 230 nm. (a) The crude linear precursor 2a with the side chain protecting group; (b) The crude LC of cyclization of 2a with the side chain protecting group; (c) The crude linear precursor of 2b without the side chain protecting group; (d) The crude LC of cyclization of 2b without the side chain protecting group. | |
To establish the optimal reaction conditions for macrocyclization. the linear peptide 2a was cyclized by using different coupling reagents for the activation of the C-terminal acid (Table 1). Unfortunately, all attempts to cyclize 2a failed to provide the desired product, but yielded the head-to-tail cyclodimerized peptide 1a (entries 1–3). The HATU-mediated coupling of 2a (entry 1) provided peptide 1a with 45% yield (based on LC—MS analysis). Cyclization with PyBOP under highly diluted conditions over several days afforded 1a with 34% yield (entry 2). Macrocyclization of 2a was performed by HATU and N-hydroxybenzotriazole (HOBT) as coupling reagent provided 1a with 27% yield (entry 3).
|
|
Table 1 Coupling conditions of peptides 2a and 2b. |

{kind=link}
{kind=link}
{kind=link}
Since cyclization with peptide 2a failed to give the desired product, we turned our attention to the unprotected peptide 2b. Three experiments were designed to examine the influence of the missing tryptophan side chain protecting group and coupling reagents. We tried various coupling reagents respectively (entries 4–6). The attempted HATU-mediated macrocyclization of 2b (entry 4) provided desired cyclic peptide 1 with 54% conversion yield and 1b with 36% conversion yield. By employing PyBOP to cyclize 2b also resulted in low yield (54% yield). The combination of HATU and HOBT as coupling reagent gave the highest yield of cyclic peptide 1 (80%) and significantly less 1b (15%) (Fig. 1d). The total conversion of the linear precursor to the corresponding cyclopeptide was determined by LC—MS analysis. To perform the cyclization of 2b, the linear peptide was dissolved in DCM; then HATU, HOBT and DIPEA dissolved in DCM was added and stirred at 25 ℃ overnight.
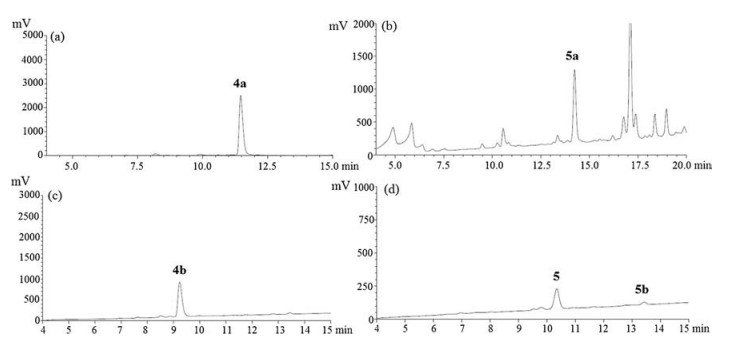
Since we found the linear peptide 2a with the side chain protecting group was unable to be cyclized to peptide 1 (Fig. 1b), while the linear peptide 2b without the side chain protecting group could be cyclized successfully with high yield (Fig. 1d), we proposed that it was the steric effect of the side chain Trp (Boc) that led to the unsuccessful cyclization. We therefore designed comparative experiment with linear peptide 4a and 4b.
We got the same experimental results as reniochalistatin E and its conformational isomers's synthesis. No desired cyclic peptide was obtained when cyclized with the peptide sequence with side chain (tBu-) in Fig. 2b. In contrast, Macrolactamization of 4b using HATU and HOBT as coupling reagent gave the highest conversion yield of cyclic peptide 5 (75%) with 16% of 5b (Fig. 2d).

|
Download:
|
| Fig. 2. LC—MS analysis for peptide 4a and 4b and their cyclization; UV was recorded at 230 nm. (a) The crude linear precursor 4a with the side chain protecting group; (b) The crude LC of cyclization of 4a; (c) The crude linear precursor of 4b without the side chain protecting group; (d) The crude LC of cyclization of 4b. | |
{kind=link}
For macrolactamization of the linear peptides without the side chain protecting group, we obtained the cyclic peptide 1 with 32% isolation yield. However, when we analyzed the NMR data (see Supporting information) of the purified compound 1, the data indicated reniochalistatin E and its conformational isomers were synthesized. The presence of Pro in the polypeptide chain controls the conformation of the cyclization, leading to the appearance of multiple isoforms [20]. Heating compound 1 at 120 ℃ overnight changed the NMR data partially but did not result in a single isoform (see Supporting information).
Also, HPLC analysis with a chiral column indicated that there were several isomers in compound 1, and the result is shown in Supporting information.
We isolated all the seven peaks (except the peak at 3.165 min, which was the solvent peak) in the figure above using a chiral preparative column, and LC–MS analysis of these products showed that all these products have the molecular weight of compound 1. We isolated the main peak at 4.973 min (compound 1) for NMR (see Supporting information) and found that it was also a mixture, which means the chiral preparative column did not yield any individual isomer.
In conclusion, we successfully synthesized proposed reniochalistatin E and its conformational isomers with 32% total yield starting from SPPS. It is of note that the side chain protecting group had a great influence on the yield of the target compound in the cyclization.
AcknowledgmentsThis work is supported by the National Natural Science Foundation of China (No. 21372183), and Program for Innovative Teams of Outstanding Young and Middle–aged Researchers in the Higher Education Institutions of Hubei Province (No. T201702).
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.05.033.
| [1] |
T. Doi, S. Kamioka, S. Shimazu, T. Takahashi, Org. Lett. 10 (2008) 817-819. DOI:10.1021/ol702965r |
| [2] |
H.M. Geng, Q.Y. Zong, J. You, et al., Sci. China Chem. 59 (2016) 293-302. DOI:10.1007/s11426-015-5477-8 |
| [3] |
K. Burgess, D. Lim, C.I. Martinez, Angew. Chem. Int. Ed. 35 (1996) 1077-1078. |
| [4] |
C. Thompson, M. Ge, D. Kahne, J. Am. Chem. Soc. 121 (1999) 1237-1244. DOI:10.1021/ja983504u |
| [5] |
M.B. Choussy, L. Neuville, R. Beugelmans, J.P. Zhu, J. Org. Chem. 61 (1996) 9309-9322. DOI:10.1021/jo961412m |
| [6] |
X.Y. Wu, J.L. Stockdill, P. Wang, S.J. Danishefsky, J. Am. Chem. Soc. 132 (2012) 4098-4100. |
| [7] |
H.Y. Lam, Y.F. Zhang, H. Liu, et al., J. Am. Chem. Soc. 135 (2013) 6272-6279. DOI:10.1021/ja4012468 |
| [8] |
K. Jin, I.H. Sam, K.H.L. Po, et al., Nat. Commun. 7 (2016) 12394-12399. DOI:10.1038/ncomms12394 |
| [9] |
A.M. Giltrap, L.J. Dowman, G. Nagalingam, et al., Org. Lett. 18 (2016) 2788-2791. DOI:10.1021/acs.orglett.6b01324 |
| [10] |
P. Desai, S.S. Pfeiffer, D.L. Boger, Org. Lett. 5 (2003) 5047-5050. DOI:10.1021/ol036083g |
| [11] |
K.X. Zhan, W.H. Jiao, F. Yang, et al., J. Nat. Prod. 77 (2014) 2678-2684. DOI:10.1021/np5006778 |
| [12] |
A. Fatino, G. Baca, C. Weeramange, et al., J. Nat. Prod. 80 (2017) 3234-3240. DOI:10.1021/acs.jnatprod.7b00656 |
| [13] |
R.B. Merrifield, J. Am. Chem. Soc. 85 (1963) 2149-2154. DOI:10.1021/ja00897a025 |
| [14] |
H.K. Cui, Y. Guo, Y. He, et al., Angew. Chem. Int. Ed. 52 (2013) 9558-9562. DOI:10.1002/anie.v52.36 |
| [15] |
C.M. Zhang, J.X. Guo, L. Wang, et al., Chin. Chem. Lett. 22 (2011) 631-634. DOI:10.1016/j.cclet.2010.12.039 |
| [16] |
J. Chen, B. Zhang, C. Xie, Y. Lu, W. Wu, Chin. Chem. Lett. 21 (2010) 391-394. DOI:10.1016/j.cclet.2009.11.026 |
| [17] |
P.J. Knerr, W.A. Donk, J. Am. Chem. Soc. 135 (2013) 7094-7097. DOI:10.1021/ja4014024 |
| [18] |
H. Zheng, F. Wang, Q. Wang, J.M. Gao, J. Am. Chem. Soc. 133 (2011) 15280-15283. DOI:10.1021/ja205911n |
| [19] |
Z.M. Wu, S.Z. Liu, X.Z. Cheng, X.R. Zhao, H.F. Hong, Chin. Chem. Lett. 28 (2017) 553-557. DOI:10.1016/j.cclet.2016.11.001 |
| [20] |
E.K. Singh, D.M. Ramsey, S.R. McAlpine, Org. Lett. 14 (2012) 1198-1201. DOI:10.1021/ol203290n |
| [21] |
K. Barlos, D. Gatos, J. Kallitsis, et al., Tetrahedron Lett. 30 (1989) 3943-3946. DOI:10.1016/S0040-4039(00)99290-6 |