 2018, Vol. 29
2018, Vol. 29 


 ,
Jun Guoa
,
Jun Guoa
b Jiangxi Key Laboratory for Mass Spectrometry and Instrumentation, East China University of Technology, Nanchang 330013, China
Native chemical ligation (NCL), initially developed by Kent and co-workers [1], is one of the most effective methods in chemical synthesis of proteins [2, 3]. It involves the selective ligation of an unprotected peptide bearing C-terminal thioester and an unprotected peptide bearing N-terminal cysteine or surrogate. During the assembly of peptide fragments, the NCL strategy is restricted in requiring a cysteine residue or a thiol auxiliary at the ligation junction. Alternatively, researchers also developed the strategies of direct aminolysis using the peptide bearing C-terminal activated oxo-esters [4], thio-esters [5] or thioacids [6] (Scheme 1), which may circumvent the restriction of thiol-assistant in NCL. However, the reactions of direct aminolysis always suffered from some limitations: low ligation rate or yield, partial epimerization at the jointed position, or the need of additive to mediate the reaction.

|
Download:
|
| Scheme 1. Cysteine-free ligation strategy for peptide synthesis (X=S or O). | |
Compared to a thiolate, the corresponding selenolate is a more nucleophilic group and a better leaving-group. Therefore, selenolderivatives [7] and selenoesters [8] were flourishingly explored in peptide ligation chemistry in recent years. Our group has successfully employed the direct aminolysis strategy of seleno-/ thio-/oxo-ester in protein modification, glycopeptide and vaccine synthesis [9]. Motivated by these recent advances, we sought a typical ligation strategy for the assembly of large (glyco)peptide without auxiliary, additive or N-cysteine residue. In this work, the direct aminolysis of peptide selenoesters was investigated for peptide ligation. It proved that no additive was required in the reaction system, and no epimerization was found at the ligation site. Furthermore, the technique was applied to assemble the glycopeptide of MUC1 mucin.
Initially, we compared the reactivity of different leaving groups in the direct aminolysis reaction. For this purpose, different esters (1a-d) (selenoester, thioester or oxoester) and the glycine amine 2a served as model substrates (Table 1). The reaction was carried out in a binary solution system of DMF and HEPES (4-(2- hydroxyethyl) piperazine-1-ethanesulfonic acid) buffer, and the process was monitored by HPLC. To our delight, for the selenoester 1a, the yield of product 3a reached 96% within 2 h (Table 1, entry 1), which demonstrated the high reactivity of 1a. Meanwhile, for the thioester 1b, the reaction proceeded slowly and the yield of the amide product 3a reached a moderate yield of 68% within 48 h (Table 1, entry 2). In contrast, the reactivity of the benzyl thioester 1c and oxoester 1d was too low to accommodate the peptide ligation (less than 5% yield at 60 h, Table 1, entries 3-4). Therefore, the selenoester 1a was the optimal activated ester in the direct aminolysis reaction.
|
|
Table 1 Aminolysis of different esters.a |

Reaction conditions were also investigated to improve the reaction efficiency. The results showed that the buffer system of DMF/HEPES (4:1, v/v, 1 mol/L HEPES containing 6 mol/L Gn·HCl, final pH 7.5) was applicable for the direct aminolysis reactions, and the satisfied yields were obtained for 10mmol/L substrates (Table S1 in Supporting information).
With optimalconditions in hand, we began toexamine whether the epimerization occurred or not at the ligation junction. The Cterminal L-Ala selenoester 1e or D-Ala selenoester 1f reacted with peptide 2b, respectively. Based on the HPLC analysis, the reactant peptides 1e and 1f, the ligation products 3b and 3c are distinguishable from each other (Fig. 1). Only one peak of the ligation product was observed from each ligation reaction, indicating no epimerization at the ligation site.

|
Download:
|
| Fig. 1. HPLC analysis of the crude peptides 1e (a), 1f (b), co-injection of the two peptides 1e and 1f (c), the ligationproducts 3b (d), 3c (e), and co-injection of the two products 3b and 3c (f). | |
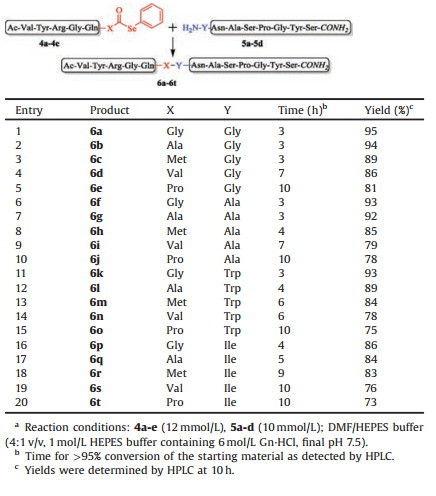
Next, we were interested in assessing the efficiency of this strategy to assemble the peptide segments. A range of peptide selenoesters 4a-e with a series of C-terminal residues (glycine, alanine, methionine, valine and proline) were synthesized (67%- 84% isolated yields, Figs. S4-S8 in Supporting information). Peptides 5a-d with a series of N-terminal residues (glycine, alanine, tryptophan and isoleucine) were also obtained by Fmoc SPPS (93%-95% isolated yields, Figs. S9-S12 in Supporting information). Then, selenoesters 4a-e reacted with N-terminal peptides 5a-d respectively (Table 2). These ligated products were purified by HPLC and confirmed by ESI-MS analysis (Figs. S13-S32 in Supporting information).
|
|
Table 2 Scope of selenoester aminolysis ligation.a |
{kind=link}
{kind=link}
To our delight, the aminolysis ligation proceeded efficiently for most peptides (Table 2). Notably, for the peptides of N-terminus bearing the glycine residue, the ligation reactions were very efficient, giving the ligation products in excellent yields (6a-e, 81%-95%, Table 2, entries 1-5). The aminolysis reaction yields were also high for the ligations with the peptides of N-terminus bearing alanine residue (6f-j, 75%-93%, Table 2, entries 6-10) and tryptophan residue (6k-o, 73%-93%, Table 2, entries 11-15), and only slightly diminished for the peptides of N-terminus containing isoleucine residue (6p-t, 73%-86%, Table 2, entries 16-20). Specially, there are still moderate yields of ligation products (6i, 6j, 6n, 6o, 6s, 6t, 73%- 79%, Table 2, entries 9, 10, 14, 15, 19, 20) when both C-terminus and N-terminus of peptides containing the sterically hindered residues (such as proline, valine, isoleucine and tryptophan).
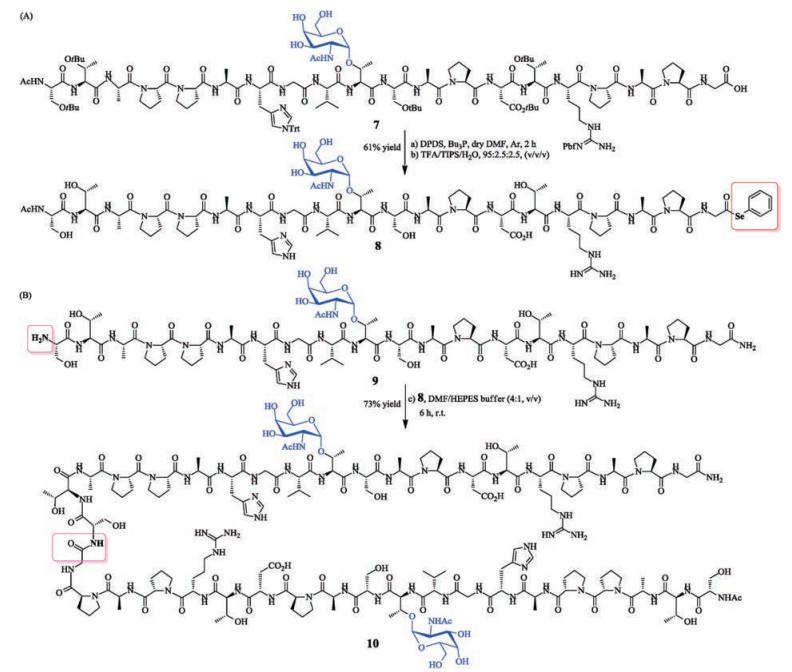
The synthesis of homogeneous glycopeptide and glycoprotein are important in biomedical study and application. MUC1 mucin are glycoproteins abundantly expressed on various cell surfaces of human adenocarcinomas (such as breast, ovarian cancers and epithelial malignancies) [10]. Therefore, MUC1 is regarded as an attractive target for immunotherapies [11]. Herein, we extended this approach of selenoester aminolysis to synthesize a glycosylated 40-mer segments of the MUC1 glycoprotein [5c, 12], which consist of the various number of tandem repeated peptide sequence of glycosylated 20 amino acids STAPPAHGVT(α-GalNAc)SAPDTRPAPG.
MUC1 glycopeptide 7 was synthesized according to the general Fmoc SPPS procedure: after assembling the protected glycopeptide chains on 2-chlorotrityl chloride resin, the acetyl protecting groups of α-GalNAc(Ac)3 were removed by the treatment of 65% hydrazine in methanol and DMF, the protected peptide 7 (Fig. S33 in Supporting information) was released from the resin using 1 vol% TFA in DCM. Glycopeptide 7 was selenoesterified, subsequently deprotected via the treatment of TFA/TIS/H2O (95:2.5:2.5, v/v/v), the desired 20-mer MUC1 selenoester 8 was obtained in 61% isolated yield (Scheme 2A, Fig. S34 in Supporting information). Similarly, the fully deprotected glycopeptide 9 of N-terminus was synthesized according to the general SPPS procedure on Rink Amide AM resin (92% isolated yield, Fig. S35 in Supporting information). Glycopeptide selenoester 8 and glycopeptide 9 were conjugated under the optimal condition of aminolysis reaction. The reaction accomplished within 6 h, and the isolated yield of desired 40-mer of glycosylated MUC1 10 reached 73% yield (Scheme 2B, Fig. S36 in Supporting information). The successful synthesis of glycopeptide MUC1 is encouraging us to investigate its application in the synthetic anti-cancer vaccine targeting MUC1 mucin using our vaccine strategy [9d, f].

|
Download:
|
| Scheme 2. Synthesis of a 40-mer MUC1 repeat glycopeptide via the direct aminolysis of selenoester. | |
{kind=link}
In conclusion, the direct aminolysis of selenoester without coupling additives was applied to assemble (glyco)peptides effectively. The ligation reactions of selenoester aminolysis featured high reaction rates in aqueous phase, and provided ligation products in excellent yields without epimerization. Furthermore, this strategy can be extended to synthesize complex (glyco)peptides/proteins.
AcknowledgmentsThis work is supported by the National Key Research and Development Program of China (No. 2017YFA0505200), the National Natural Science Foundation of China (No. 21772056), Jiangxi Key Laboratory for Mass Spectrometry and Instrumentation Open Foundation (No. JXMS201701), the self-determined research funds of CCNU from the colleges' basic research and operation of MOE (No. CCNU18TS011) and the Program of Introducing Talents of Discipline to Universities of China (111 program, No. B17019).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.04.016.
| [1] |
P.E. Dawson, T.W. Muir, I. Clark-Lewis, S.B. Kent, Science 266 (1994) 776-779. DOI:10.1126/science.7973629 |
| [2] |
(a) C. P. Hackenberger, D. Schwarzer, Angew. Chem. Int. Ed. 47 (2008) 10030-10074; (b) C. Unverzagt, Y. Kajihara, Chem. Soc. Rev. 42 (2013) 4408-4420; (c) C. T. Wong, C. L. Tung, X. Li, Mol. Biosyst. 9 (2013) 826-833; (d) H. Hojo, Curr. Opin. Struct. Boil. 26 (2014) 16-23; (e) L. R. Malins, R. J. Payne, Aust. J. Chem. 68 (2015) 521-537; (f) Y. C. Huang, G. M. Fang, L. Liu, Natl. Sci. Rev. 3 (2016) 107-116; (g) S. Bondalapati, M. Jbara, A. Brik, Nat. Chem. 8 (2016) 407; (h) H. Li, S. Dong, Sci. China Chem. 60 (2017) 201-213; (i) V. Agouridas, O. El Mahdi, M. Cargoët, O. Melnyk, Bioorg. Med. Chem. 25 (2017) 4938-4945; (j) J. Yang, J. Zhao, Sci. China Chem. 61 (2018) 97-112; (k) S. Kent, Bioorg. Med. Chem. 25 (2017) 4926-4937. |
| [3] |
(a) J. B. Blanco-Canosa, B. Nardone, F. Albericio, P. E. Dawson, J. Am. Chem. Soc. 137 (2015) 7197-7209; (b) Z. Wang, W. Xu, L. Liu, T. F. Zhu, Nat. Chem. 8 (2016) 698-704; (c) Y. Gui, L. Qiu, Y. Li, H. Li, S. Dong, J. Am. Chem. Soc. 138 (2016) 4890-4899; (d) A. M. Levinson, J. H. McGee, A. G. Roberts, et al., J. Am. Chem. Soc. 139 (2017) 7632-7639; (e) S. Tang, C. Zuo, D. L. Huang, et al., Nat. Protoc. 12 (2017) 2554-2569; (f) H. Liu, Y. Zhang, R. Wei, G. Andolina, X. Li, J. Am. Chem. Soc. 139 (2017) 13420-13428; (g) K. Jin, T. Li, H. Y. Chow, H. Liu, X. Li, Angew. Chem. 129 (2017) 14799-14803; (h) L. J. Liang, Y. Si, S. Tang, et al., Chin. Chem. Lett. 29 (2018) 1155-1159. |
| [4] |
(a) D. Kemp, Z. W. Bernstein, G. N. McNeil, J. Org. Chem. 39 (1974) 2831-2835; (b) D. Kemp, S. L. H. Choong, J. Pekaar, J. Org. Chem. 39 (1974) 3841-3847; (c) G. Chen, Q. Wan, Z. Tan, et al., Angew. Chem Int. Ed. 46 (2007) 7383-7387; (d) C. Xu, J. Xu, H. Liu, X. Li, Chin. Chem. Lett. 29 (2018) 1119-1122. |
| [5] |
(a) J. Blake, Int. J. Pept. Protein Res. 17 (1981) 273-274; (b) S. Aimoto, Biopolymers 51 (1999) 247-265; (c) R. J. Payne, S. Ficht, W. A. Greenberg, C. H. Wong, Angew. Chem. Int. Ed. 47 (2008) 4411-4415; (d) C. L. Tung, C. T. T. Wong, X. Li, Org. Biomol. Chem. 13 (2015) 6922-6926. |
| [6] |
(a) D. Crich, I. Sharma, Angew. Chem. Int. Ed. 48 (2009) 2355-2358; (b) P. Wang, S. J. Danishefsky, J. Am. Chem. Soc. 132 (2010) 17045-17051; (c) S. M. Mali, S. V. Jadhav, H. N. Gopi, Chem. Commun. 48 (2012) 7085-7087. |
| [7] |
(a) M. D. Gieselman, L. Xie, W. A. Van Der Donk, Org. Lett. 3 (2001) 1331-1334; (b) R. Quaderer, A. Sewing, D. Hilvert, Helv. Chim. Acta 84 (2001) 1197-1206; (c) R. J. Hondal, B. L. Nilsson, R. T. Raines, J. Am. Chem. Soc. 123 (2001) 5140-5141; (d) N. Metanis, E. Keinan, P. E. Dawson, Angew. Chem. Int. Ed. 49 (2010) 7049-7053; (e) S. Dery, P. S. Reddy, L. Dery, R. Mousa, R. N. Dardashti, N. Metanis, Chem. Sci. 6 (2015) 6207-6212; (f) L. R. Malins, N. J. Mitchell, S. McGowan, R. J. Payne, Angew. Chem. Int. Ed. 54 (2015) 12716-12721. |
| [8] |
(a) T. Durek, P. F. Alewood, Angew. Chem. Int. Ed. 50 (2011) 12042-12045; (b) A. Ghassemian, X. Vila-Farrés, P. F. Alewood, T. Durek, Bioorg. Med. Chem. 21 (2013) 3473-3478; (c) L. R. Malins, N. J. Mitchell, R. J. Payne, J. Pept. Sci. 20 (2014) 64-77; (d) M. Raj, H. Wu, S. L. Blosser, M. A. Vittoria, P. S. Arora, J. Am. Chem. Soc. 137 (2015) 6932-6940; (e) L. Raibaut, H. Drobecq, O. Melnyk, Org. Lett. 17 (2015) 3636-3639; (f) N. J. Mitchell, L. R. Malins, X. Liu, etal., J. Am. Chem. Soc. 137 (2015)14011-14014; (g)A. Temperini, F. Piazzolla, L. Minuti, M. Curini, C. Siciliano, J. Org. Chem. 82 (2017) 4588-4603; (h) C. C. Hanna, S. S. Kulkarni, E. E. Watson, B. Premdjee, R. J. Payne, Chem. Commun. 53 (2017) 5424-5427; (i) N. J. Mitchell, J. Sayers, S. S. Kulkarni, et al., Chemistry 2 (2017) 703-715; (j) T. Takei, T. Andoh, T. Takao, H. Hojo, Angew. Chem. 129 (2017) 15914-15917. |
| [9] |
(a) X. F. Gao, J. J. Du, Z. Liu, J. Guo, Org. Lett. 18 (2016) 1166-1169; (b) J. J. Du, X. F. Gao, L. M. Xin, et al., Org. Lett. 18 (2016) 4828-4831; (c) X. G. Yin, X. F. Gao, J. J. Du, et al., Org. Lett 18 (2016) 5796-5799; (d) X. G. Yin, X. Z. Chen, W. M. Sun, et al., Org. Lett. 19 (2017) 456-459; (e) J. Wang, R. Y. Zhang, Y. C. Wang, et al., Synlett 28 (2017) 1934-1938; (f) Z. Liu, J. Guo, Carbohydr. Res. 452 (2017) 78-90. |
| [10] |
(a) Taylor-Papadimitriou, J. Burchell, D. Miles, M. Dalziel, Biochim. Biophys. Acta 1455 (1999) 301-313; (b) V. Apostolopoulos, L. Stojanovska, S. E. Gargosky, Cell. Mol. Life Sci. 72 (2015) 4475-4500. |
| [11] |
(a) Z. H. Huang, L. Shi, J. W. Ma, et al., J. Am. Chem. Soc. 134 (2012) 8730-8733; (b) N. Gaidzik, U. Westerlind, H. Kunz, Chem. Soc. Rev. 42 (2013) 4421-4442; (c) H. Cai, Z. Y. Sun, M. S. Chen, et al., Angew. Chem. Int. Ed. 53 (2014) 1699-1703; (d) M. Movahedin, T. M. Brooks, N. T. Supekar, et al., Glycobiology 27 (2017) 677-687. |
| [12] |
C. Xu, H.Y. Lam, Y. Zhang, X. Li, Chem. Commun. 49 (2013) 6200-6202. DOI:10.1039/c3cc42573h |