 2018, Vol. 29
2018, Vol. 29 



As an important class of molecular constructs from both natural and synthetic sources, cyclic peptides attract the attention of the researchers from synthetic chemistry and medicinal chemistry. Compared with the corresponding linear peptides, cyclic peptides are prone to show improved binding affinity to protein targets and higher stability against degradation, benefiting from the restricted conformations [1-3]. Compared with the linear peptides which can be readily and even automatically assembled via solid phase peptide synthesis (SPPS), the syntheses of cyclic peptides pose additional challenges in the head-to-tail macrolactamization of the linear precursors with protected side chains [4]. These challenges include racemization of the C-terminal amino acid residues in the linear precursors and the undesired oligomerization, which have to be overcome by extensive optimization of the coupling conditions (reagents, bases, concentrations, etc.), variation of the cyclization sites, and even incorporation of unnatural structure units [5].
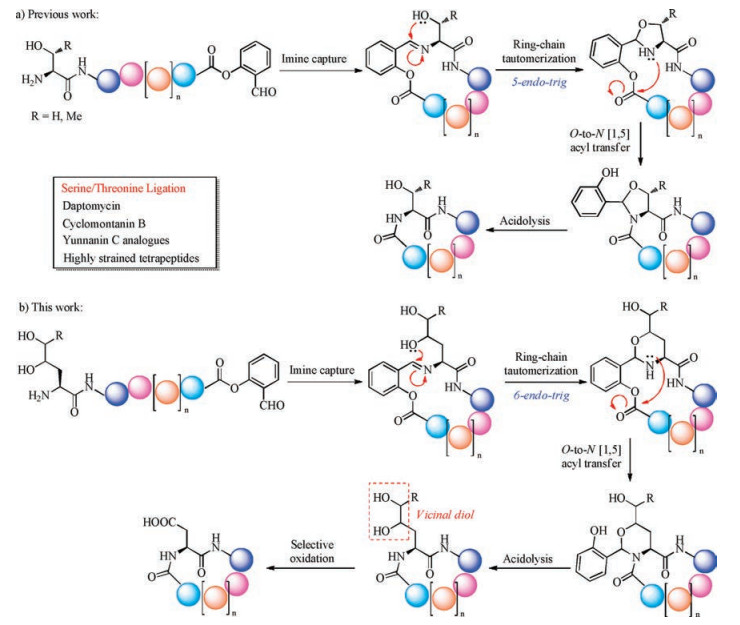
As an alternative strategy to the traditional head-to-tail peptide cyclization based on coupling, the chemical peptide ligation with the feature of site specific formation of the peptide linkage between side chain unprotected fragments without racemization have been adapted to the synthesis of cyclic peptides [6]. Since the first publication of native chemical ligation by Kent et al. in 1994 [7], several peptide ligation strategies, including native chemical ligation (NCL) [8-12], pseudoproline ligation [13, 14], α-ketoacidhydroxylamine (KAHA) ligation [15-17], traceless Staudinger ligation [18], 2-formylthiophenol ligation [19] and enzyme (subtiligase [20], butelase [21-23], sortase [24]) catalyzed ligations, have been used in the synthesis of cyclic peptides and even cyclic proteins. In 2010, our group developed a novel peptide ligation, termed serine threonine ligation (STL), between the peptide C-terminal salicylaldehyde esters and peptides with Nterminal serine or threonine residues (Scheme 1a) [25-30]. This ligation works through the imine capture between the two peptide fragments, followed by the side chain nucleophile (hyroxyl group in Ser or Thr) triggered ring-chain tautomerization and subsequent O-to-N [1, 5] acyl transfer. The so-obtained N, O-benzylidene acetal is acid labile, which can be easily cleaved under mild condition to give the product with native peptide linkage. This new ligation has been applied in the synthesis of linear peptide drugs [31], proteins with post-translational modifications [32-34], as well as cyclic peptides/depsipeptides [35] (daptomycin [36, 37], teixobactin [38, 39], yunnanin C analogues [40], cyclomontanin B [41]) with variable ring sizes (from highly strained cyclic tetrapeptides [42, 43] to highly flexible decapeptides). As an extension of our former work, we conceived of a new ligation method for peptide cyclization, based on the analogous side chain nucleophile triggered ring-chain tautomerization and O-to-N acyl transfer. As illustrated in Scheme 1b, we designed an aspartic acid ligation, facilitated by the γ-amino alcohol based ligation and oxidation. Compared with the serine/threonine ligation, the imine capture and the O-to-N acyl transfer are in common, while the side chain nucleophile triggered ring-chain tautomerization works in a 6- endo-trig manner rather than a 5-endo-trig manner. To facilitate further transformation, vicinal diol functionality is introduced onto the N-terminal residue instead of a single hydroxyl group, which can be cleaved under mild oxidation condition to give a carboxylic acid residue. Through this process, a native aspartic acid residue will be formed at the ligation site. Herein, we would like to document how we develop this ligation and apply it to cyclic peptide synthesis.

|
Download:
|
| Scheme 1. Design of the aspartic acid ligation for peptide cyclization facilitated by γ-amino alcohol based ligation and oxidation. | |
{kind=link}
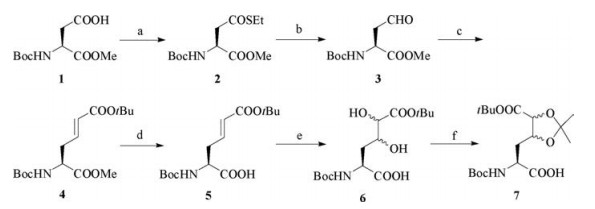
To test our idea, we designed a Boc protected dihydroxylated amino acid building block (abrreviated as Dh) 7 for solid phase peptide synthesis, which contains the vicinal diol functionality in the side chain. As illustrated in Scheme 2, the synthesis started from commercial available Boc protected L-aspartic acid monomethyl ester 1, which was transformed into side chain thioester 2. After the Fukuyama reduction [44], the aldehdye 3 was directly trapped by phosphoniumylide to give alkene 4. The methyl ester in 4 was selectively hydrolyzed in the presence of tert-butyl ester under mild condition, then the vicinal diol functionality was installed on 5 via osmium(Ⅷ) catalyzed cis-dihydroxylation [45]. An inseparable mixture of diastereomers 6 was obtained in a moderate yield, and then the diol was further protected by acetonide to give 7. Details of this synthesis can be found in Supporting information.

|
Download:
|
| Scheme 2. Synthesis of dihydroxylated amino acid building block (Dh). a) EtSH, DCC, DMAP, DCM, 90%. b) Pd/C, Et3SiH, DCM. c) tBuOOCCH = PPh3, THF, 87% over 2 steps. d) LiOH (aq.), THF/H2O, 85%. e) 4-Methylmorpholine N-oxide monohydrate, K2OsO4·2H2O, acetone/H2O, 44%. f) 2, 2-Dimethoxypropane, BF3·OEt2, acetone, 81%. | |
{kind=link}
After obtaining the Dh building block 7, we went forward to the preparation of the peptide C-terminal salicylaldehyde (SAL) esters, the linear precursors for Ser/Thr ligation. To achieve this, the BocSPPS based method was used [42]. As illustrated in Scheme 3, from AM resin, the 2-hyroxylcinnamoyl linker (the surrogate of SAL) was installed to give 8, followed by the assembly of the peptide sequence. After finishing the desired sequence with the building block 7 installed onto the N-terminus, the resin bounded peptide 9 was subjected to the global deprotection. The SAL ester was generated from the released peptide via ozonolysis, and the product 10a-e were obtained after preparative HPLC purification in 25%-42% yields (calculated based on resin loading).

|
Download:
|
| Scheme 3. Synthesis of peptide C-terminal SAL ester with N-terminal Dh via Boc-SPPS. | |
{kind=link}
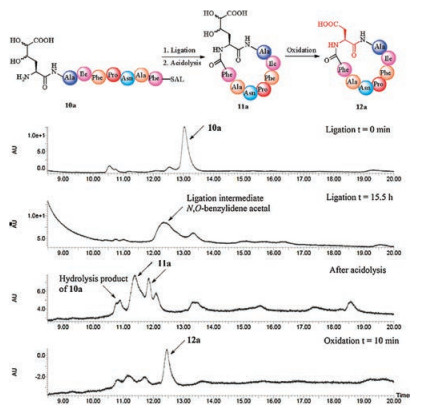
With the desired peptide SAL esters 10a-e in hand, we started to test this peptide cyclization. As an example, the peptide 10a was dissolved in the pyridine/acetic acid buffer to form a 20 mmol/L solution, and the progress of the ligation was monitored by LC-MS. After screening of the pyridine/acetic acetic buffer compositions (from 9:1 to 1:6, molar ratio), the 9:1 buffer was found to be optimal. The HPLC traces of the reaction under the optimized condition were shown in Fig. 1. The full conversion of the peptide SAL ester 10a was observed after 15.5 h, and the N, O-benzylidene acetal intermediate (broaden peak) was formed as a mixture of diastereomers (due to the mixture of Dh building block) via the expected 6-endo-trig tetrahydro-1, 3-oxazine formation and O-to-N acyl transfer. After blowing off the pyridine/acetic acid by flow gas, the crude intermediate was then treated with TFA/H2O/TIPS (95/2.5/2.5, v/v/v) cocktail to cleave the benzylidene acetal, and the desired cyclic peptide 11a with Dh at the ligation site was formed, also as mixture of diastereomers.

|
Download:
|
| Fig. 1. HPLC monitoring of the aspartic acid ligation facilitated peptide cyclization. | |
{kind=link}
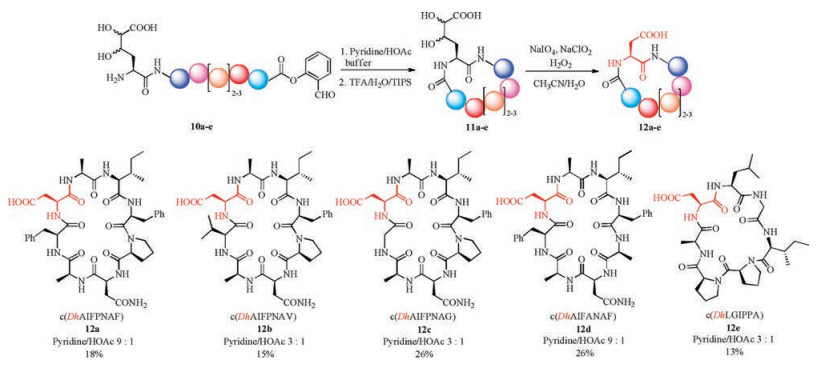
To achieve the desired aspartic acid ligation, we optimized the site-selective oxidation of the diol side chain of Dh residue. After extensive screening of the reagents and conditions (details see Supporting information), to our delight, the formation of the aspartic acid from Dh residue was realized in one-pot manner under the treatment of the cyclic peptide 11a by the mixture of NaIO4, NaClO2 and H2O2 in MeCN/H2O. As an example, the crude ligation products 11a (1.0 equiv.) was mixed with NaIO4 (15.0 equiv.) and NaClO2 (10.0 equiv.) in CH3CN/H2O (1/1, v/v) at room temperature. After the addition of H2O2 (50 wt% in H2O, 20.0 equiv.), gas was evolved and a white precipitate formed in the stirred yellow solution. The oxidation reaction was completed within 10 min, as indicated by LC-MS monitoring. The product 12a was obtained in 18% yield over 3 steps (calculated based on the purified SAL ester 10a) after HPLC purification and lyophilization.
To evaluate the scope of this new peptide cyclization method, we tested the reaction on a series of peptide SAL esters with varied sequences and chain length. As shown in Scheme 4, for substrates 10a-e, different pyridine/acetic acid ratios (3:1 or 9:1) were found to be optimal for the ligation step. Pyridine was used as the dominant composition in general, which was in accordance with our former observations in cyclic tetrapeptide synthesis via serine/ threonine ligation [42]. Val or less bulky Gly were tolerated. For the peptide 10d without turn-inducing Pro residue, longer reaction time (up to 4 days) was needed to achieve full conversion. After the acidolysis of the N, O-benzylidene acetal intermediates, the crude cyclic peptides 11a-e were subjected to the optimized oxidation condition to give the octapeptides 12a-d and heptapeptide 12e in 13%-26% yields after HPLC purification.

|
Download:
|
| Scheme 4. Substrate scope of the aspartic acid ligation facilitated peptide cyclization. | |
{kind=link}
In conclusion, we developed a novel aspartic acid ligation derived from serine/threonine ligation, in which the γ-amino alcohol based ligation worked through a ring-chain tautomerization via 6-endo-trig process and subsequent O-to-N [1, 5] acyl transfer. The oxidative cleavage of the diol functionality into carboxylic acid was realized by NaIO4/NaClO2/H2O2 in water containing system in one-pot manner. The method was successfully used in the synthesis of cyclic peptides with aspartic acid residue in the sequences. Nevertheless, this γ-amino alcohol based ligation was less efficient as compared to the serine/threonine ligation. Both ligation methods proceed through ring-chain tautomerization step and acyl transfer step. According to the Baldwin empirical rule, the γ-amino alcohol based ligation giving a 6-endo-trig ring-chain tautomerization is more favored than the 5-endo-trig cyclization from serine/threonine ligation using β-amino alcohol. Thus, it is very likely that, during the acyl transfer step, the 5-6 fused bicyclic transition state in serine/threonine ligation is much more favored than the 6-6 fused bicyclic transition state in the γ-amino alcohol based ligation. The current effort to apply for this ligation in protein chemical synthesis is ongoing in our laboratory.
AcknowledgmentsThis work was supported by the Research Grants Council (Nos. 17309616, C6009-15G) of Hong Kong, The National Science Foundation of China (Nos. 21672180, 91753101), the Area of Excellence Scheme of the University of Grants Committee of Hong Kong (No. AoE/P-705/16).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.03.012.
| [1] |
Y.S. Ong, L. Gao, K.A. Kalesh, et al., Curr. Top. Med. Chem. 17 (2017) 2302-2318. |
| [2] |
A.T. Bockus, C.M. McEwen, R.S. Lokey, Curr. Top. Med. Chem. 13 (2013) 821-836. DOI:10.2174/1568026611313070005 |
| [3] |
S.H. Joo, Biomol. Ther. 20 (2012) 19-26. DOI:10.4062/biomolther.2012.20.1.019 |
| [4] |
C.J. White, A.K. Yudin, Nat. Chem. 3 (2011) 509-524. DOI:10.1038/nchem.1062 |
| [5] |
Z.M. Wu, S.Z. Liu, X.Z. Chen, et al., Chin. Chem. Lett. 27 (2016) 1731-1739. DOI:10.1016/j.cclet.2016.04.024 |
| [6] |
G.K.T. Nguyen, C.T.T. Wong, J. Biochem. Chem. Sci. 2017 (2017) 1-13. |
| [7] |
P.E. Dawson, T.W. Muir, I. Clark-Lewis, et al., Science 266 (1994) 776-779. DOI:10.1126/science.7973629 |
| [8] |
P.E. Dawson, S.B.H. Kent, Annu. Rev. Biochem. 69 (2000) 923-960. DOI:10.1146/annurev.biochem.69.1.923 |
| [9] |
G.M. Fang, Y.M. Li, F. Shen, et al., Angew. Chem. Int. Ed. 50 (2011) 7645-7649. DOI:10.1002/anie.201100996 |
| [10] |
S. Tang, J.S. Zheng, K. Yang, et al., Acta Chim. Sin. 70 (2012) 1471-1476. DOI:10.6023/A12040166 |
| [11] |
J.X. Wang, G.M. Fang, Y. He, et al., Angew. Chem. Int. Ed. 54 (2015) 2194-2198. DOI:10.1002/anie.201408078 |
| [12] |
K. Jin, T. Li, H.Y. Chow, et al., Angew. Chem. Int. Ed. 56 (2017) 14607-14611. DOI:10.1002/anie.201709097 |
| [13] |
C.F. Liu, J.P. Tam, J. Am. Chem. Soc. 116 (1994) 4149-4153. DOI:10.1021/ja00089a001 |
| [14] |
P. Botti, T.D. Pallin, J.P. Tam, J. Am. Chem. Soc. 118 (1996) 10018-10024. DOI:10.1021/ja954278g |
| [15] |
J.W. Bode, Acc. Chem. Res. 50 (2017) 2104-2115. DOI:10.1021/acs.accounts.7b00277 |
| [16] |
F. Rohrbacher, G. Deniau, A. Luther, et al., Chem. Sci. 6 (2015) 4889-4896. DOI:10.1039/C5SC01774B |
| [17] |
T. Fukuzumi, L. Ju, J.W. Bode, Org. Biomol. Chem. 10 (2012) 5837-5844. DOI:10.1039/c2ob25129a |
| [18] |
R. Kleineweischede, C.P.R. Hackenberger, Angew. Chem. Int. Ed. 47 (2008) 5984-5988. DOI:10.1002/anie.200801514 |
| [19] |
C.L. Tung, C.T.T. Wong, X. Li, Org. Biomol. Chem. 13 (2015) 6922-6926. DOI:10.1039/C5OB00825E |
| [20] |
D.Y. Jackson, J.P. Burnier, J.A. Wells, J. Am. Chem. Soc. 117 (1995) 819-820. DOI:10.1021/ja00107a027 |
| [21] |
G.K.T. Nguyen, S. Wang, Y. Qiu, et al., Nat. Chem. Biol. 10 (2014) 732-738. DOI:10.1038/nchembio.1586 |
| [22] |
G.K.T. Nguyen, Y. Qiu, Y. Cao, et al., Nat. Protoc. 11 (2016) 1977-1988. DOI:10.1038/nprot.2016.118 |
| [23] |
G.K.T. Nguyen, X. Hemu, J.P. Quek, et al., Angew. Chem. Int. Ed. 55 (2016) 12802-12806. DOI:10.1002/anie.201607188 |
| [24] |
Z.M. Wu, S.Z. Liu, X.Z. Cheng, et al., Chin. Chem. Lett. 28 (2017) 553-557. DOI:10.1016/j.cclet.2016.11.001 |
| [25] |
X. Li, H.Y. Lam, Y. Zhang, C.K. Chan, Org. Lett. 12 (2010) 1724-1727. DOI:10.1021/ol1003109 |
| [26] |
Y. Zhang, C. Xu, H.Y. Lam, et al., Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 6657-6662. DOI:10.1073/pnas.1221012110 |
| [27] |
C.T.T. Wong, T. Li, H.Y. Lam, et al., Front. Chem. 2 (2014). DOI:10.3389/fchem.2014.00028 |
| [28] |
C.L. Lee, X. Li, Curr. Opin. Chem. Biol. 22 (2014) 108-114. DOI:10.1016/j.cbpa.2014.09.023 |
| [29] |
C.L. Lee, X. Li, Sci. China Chem. 59 (2016) 1061-1064. |
| [30] |
H. Liu, X. Li, Org. Biomol. Chem. 12 (2014) 3768-3773. DOI:10.1039/C4OB00392F |
| [31] |
Y. Zhang, T. Li, X. Li, Org. Biomol. Chem. 11 (2013) 5584-5587. DOI:10.1039/c3ob41027g |
| [32] |
C. Xu, H.Y. Lam, Y. Zhang, et al., Chem. Commun. 49 (2013) 6200-6202. DOI:10.1039/c3cc42573h |
| [33] |
T. Li, H. Liu, X. Li, Org. Lett. 18 (2016) 5944-5947. DOI:10.1021/acs.orglett.6b03056 |
| [34] |
C.L. Lee, H. Liu, C.T.T. Wong, et al., J. Am. Chem. Soc. 138 (2016) 10477-10484. DOI:10.1021/jacs.6b04238 |
| [35] |
C.L. Lee, H.Y. Lam, X. Li, Nat. Prod. Rep. 32 (2015) 1274-1279. DOI:10.1039/C5NP00001G |
| [36] |
H.Y. Lam, Y. Zhang, H. Liu, et al., J. Am. Chem. Soc. 135 (2013) 6272-6279. DOI:10.1021/ja4012468 |
| [37] |
D. Lin, H.Y. Lam, W. Han, et al., Bioorg. Med. Chem. Lett. 27 (2017) 456-459. DOI:10.1016/j.bmcl.2016.12.046 |
| [38] |
K. Jin, I.H. Sam, K.H.L. Po, et al., Nat. Commun. 7 (2016) 12394. DOI:10.1038/ncomms12394 |
| [39] |
K. Jin, K.H.L. Po, S. Wang, et al., Bioorg. Med. Chem. 25 (2017) 4990-4995. DOI:10.1016/j.bmc.2017.04.039 |
| [40] |
C.T.T. Wong, H.Y. Lam, X. Li, Tetrahedron 70 (2014) 7770-7773. DOI:10.1016/j.tet.2014.05.080 |
| [41] |
C.T.T. Wong, H.Y. Lam, X. Li, Org. Biomol. Chem. 11 (2013) 7616-7620. DOI:10.1039/c3ob41631c |
| [42] |
C.T.T. Wong, H.Y. Lam, T. Song, et al., Angew. Chem. Int. Ed. 52 (2013) 10212-10215. DOI:10.1002/anie.201304773 |
| [43] |
J.F. Zhao, X.H. Zhang, Y.J. Ding, et al., Org. Lett. 15 (2013) 5182-5185. DOI:10.1021/ol402279h |
| [44] |
T. Fukuyama, S.C. Lin, L. Li, J. Am. Chem. Soc. 112 (1990) 7050-7051. DOI:10.1021/ja00175a043 |
| [45] |
Y. Yoshimura, C. Ohara, T. Imahori, et al., Bioorg. Med. Chem. 16 (2008) 8273-8286. DOI:10.1016/j.bmc.2008.06.016 |