 2018, Vol. 29
2018, Vol. 29 



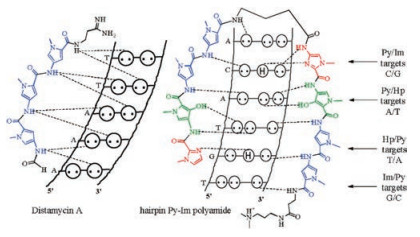
DNA is widely involved in many functional roles including replication, and transcription. A myriad of devastating health diseases such as cancer and some hereditary conditions are related to the aberrant expression of DNA [1]. DNA has become the prime target for some related diseases. Many synthetic small molecules targeting DNA via binding to the grooves, intercalation, crosslinking, or DNA strand-scission, have been discovered to disturb DNA functions [2]. N-Methylpyrrole (Py)-N-methylimidazole (Im) polyamides (PIPs), originally derived from distamycin A, are a class of programmable minor-groove binders that recognize predetermined DNA sequences with nanomolar binding affinity [3, 4]. They are consisting of two polyamide strands with a γ-aminobutyric acid (γ-turn) linkage, and form hairpin-like structures. Their specificity is achieved by forming hydrogen bonds with double-strand DNA (dsDNA) based on the WatsonCrick rules, that is antiparallel Im/Py pair discriminates G/C base pair from C/G, and N-methyl-3-hydroxypyrrole (Hp)/Py shows specificity for T/A over A/T, whereas Py/Py pair recognizes both A/T and T/A base pairs. This binding mode is oriented N→C with respect to the 5′→3′ direction of the adjacent DNA in the minor groove (Fig. 1). Once Py-Im polyamides bind with matched dsDNA, DNA-binding proteins will be competitively displaced, so that the expression of corresponding genes is inhibited or promoted. In recent years, various Py-Im polyamides and their conjugates have been developed to modulate endogenous expression of nuclear genes and proteins. Py-Im polyamides have also been used as targeting ligands for DNA recognition and bioimaging. To date, there have been many excellent reviews introducing the development of Py-Im polyamides in this field [3-7]. In this review, we summarized the new synthetic methods and the structure-activity relationship of Py-Im polyamides, and outlined the recent advances of their applications as genetic modulators and targeting ligands.

|
Download:
|
| Fig. 1. Binding models of distamycin A and hairpin Py-Im polyamide with dsDNA. | |
2. New synthetic methods for Py-Im polyamides
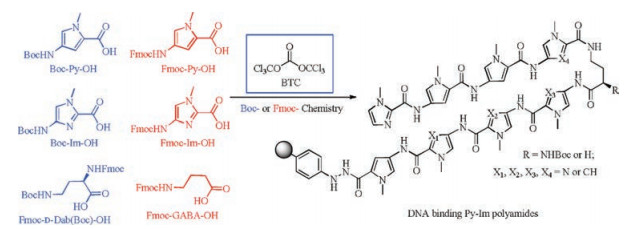
Initial Py-Im polyamide syntheses started from long and difficult solution phase. A significant advance was the solid-phase synthesis of Py-Im polyamides developed by Baird and Dervan, which reduced the synthetic timescale for a single polyamide from months to days or weeks [8, 9]. Moreover, multifunctional polyamides were produced by using Kaiser oxime or safety-catch hydrazine resin followed by nucleophilic cleavage with various compounds [10]. However, the low yields and lengthy durations were still the crucial problems in the synthesis of Py-Im polyamides [11]. Hence, Dervan et al. developed a convergent synthesis of Py-Im polyamides on gram-scale with minimal use of chromatography by using a [3 + 2+4]-peptide fragment-coupling strategy in solution phase [12]. In recent years, microwave assisted solid-phase synthesis was utilized to prepare Py-Im polyamides, which significantly enhanced the coupling yields and reduced the coupling times [13]. Burley et al. developed a convergent synthesis of Py-Im polyamides with superior yields and purities, which involved the Boc-based solid-phase peptide synthesis (SPPS) of the C-terminal fragment and the solution phase synthesis of the Nterminal fragment [14]. We previously reported a facile and highly efficient solid-phase synthesis of Py-Im polyamides using the costeffective triphosgene (BTC) as activating agent [15]. BTC converts carboxylic acids into highly electrophilic acid chlorides, which readily acylate the sterically hindered and electronically deactivated amines. However, the strong exothermic nature and the large amount of precipitates generated in the process of BTC-mediated activation were unfavorable to automated synthesis. Therefore, development of a milder and solid free activation method was crucial for the automation of the BTC strategy. We found that the insoluble collidine hydrochloride salts produced by the preactivation of the carboxyl acid with BTC and collidine in THF could be converted to the soluble N, N-diisopropylethylamine (DIEA) hydrochloride by the addition of a DIEA/DMF solution. So we used this special activation procedure to avoid the potential clogging of the peptide synthesizer pipelines, and realized the fully automated solid-phase synthesis of Py-Im polyamides, which represented a significant step forward for the multiple, parallel production of PyIm polyamides (Fig. 2) [16].

|
Download:
|
| Fig. 2. Synthesis of Py-Im polyamides via BTC activation. | |
In addition to linear and hairpin Py-Im polyamides, tandem hairpin structures have been designed and synthesized. Tandem hairpin Py-Im polyamides are comprised of two or more hairpin Py-Im polyamides linked with a flexible hinge segment, thus can recognize up to 10 base pairs. However, the synthesis of this kind of polyamides suffered from low yields because multiple steps were required to remove Boc groups or Fmoc groups in Fmoc SPPS or Boc SPPS, respectively. By preparing a new tetraamide unit in solution phase and introducing it to Fmoc SPPS, Sugiyama et al. developed an efficient synthetic route for creating tandem hairpin Py-Im polyamides [17]. Using this method, they synthesized a series of tandem hairpin Py-Im polyamides including tandem dimer [18], trimer [19] and tetramer [20].
3. Structure-activity relationship of Py-Im polyamidesStructures of Py-Im polyamides influence their activity. Compared to natural distamycin A (interacted with DNA in a 1:1 stoichiometry) (Fig. 1), hairpin Py-Im polyamides, in which a γ-turn linker connects two polyamide strands, display approximately 100-fold higher affinity [21]. A number of molecular recognition researches were conducted to evaluate the structureactivity relationship of Py-Im polyamides. For example, previous reports have demonstrated the cell uptake and nuclear localization of Py-Im polyamides were related to Py/Im content, number and location of positive and negative charges, the modification of Cterminus and N-terminus, choice of fluorophore, linker composition and attachment [22-25]. It was noted that hairpin Py-Im polyamides with eight or more rings became over-curved and would no longer match the minor groove shape of dsDNA. In replacement of Py, introduction of a flexible β-alanine (β) to longer Py-Im polyamides was needed to compensate the structural incompatibility. But β substitution could sometimes decrease the binding affinity of Py-Im polyamides with DNA. Wilson et al. found the number and position of β substitutions affected the affinity and specificity of Py-Im polyamides [26, 27]. They summarized when Py between two Im was replaced by β, the binding affinity tended to go up (such as KA2034 in Table 1), while if β appeared on a lower strand consisting of four Py, the binding affinity tended to go down (such as KA2041 and KA2114 in Table 1). They reasoned that Im heterocycles were more rigid than Py rings, and displacement Py with β increased the flexibility of the whole molecule. For the former, increase of the molecular flexibility adjusted Py-Im polyamides to the curvature of DNA more easily. Inversely for the latter, this increase reduced van der Waals interaction between Py-Im polyamides and dsDNA, leading to slack the binding.
|
|
Table 1 Equilibrium binding constants for Py-Im polyamides with β-alanine substituents [26]. |

{kind=link}
{kind=link}
According to well-defined pairing rules, Py-Im polyamides retain the orientation preferences of aligning N to C terminus with respect to the 5′-3′ direction of the DNA (referred to as the forward orientation). However, for some Py-Im polyamides, reversed binding (a C to N terminus alignment of Py-Im polyamides with respect to the 5′-3′ direction of DNA) has been observed as the dominant orientation. Dervan et al. reported β-containing polyamides with different γ-turn units (α-amino substituted, β-amino substituted or unsubstituted) displayed unanticipated binding motifs with DNA by Bind-N-Seq [28]. They discovered that conformationally flexible, β-containing polyamides with a β-amino-γ-turn linker preferred a reverse orientation, and the forward orientation was recovered when β-amino-γ-turn linker was replaced by α-amino-γ-turn linker. However, for the polyamide sequence PyImβIm-γ-PyImβIm with a reversed binging mode, replacement of one β-alanine with Py to afford PyImPyIm-γ-PyImβIm restored the preference of forward orientation. These results provide significant insights for designing rationally Py-Im polyamides with high affinity and specificity in the future.
4. The applications of Py-Im polyamides 4.1. Py-Im polyamide-mediated transcriptional modulationsPy-Im polyamides are capable of binding the minor groove of helix DNA with high affinity and sequence specificity. Following Py-Im polyamides binding to DNA, the minor groove is widened, and the major groove is bent and compressed to block transcription factors (TFs) binding, which interferes with the interactions between transcription factors and DNA, so that the expression of corresponding genes is inhibited or promoted. Previously, a number of sequence-specific DNA binding Py-Im polyamides have been successfully developed to modulate the expressions of disease related genes, such as TGF-β1 [29], NF-κB [30], ARE [31] and MMP-9 [32]. There have been some new targets inhibited by Py-Im polyamides, such as EVI1 [33], estrogen response element [34], ABCA1 [35] and E2F1 [36]. Here we summarized the new research progresses of Py-Im polyamide-mediated transcriptional modulations in the recent years.
Dervan et al. designed a cell-permeable Py-Im polyamide (ARE- 1) (Fig. 3) to target the androgen response element for inhibiting androgen-induced expression of prostate-specific antigen [31, 37]. They further found ARE-1 was active against two enzalutamideresistant xenografts [38]. Enzalutamide is a potent androgen receptor (AR) ligand-binding domain (LBD) antagonist, but can activate an alternative nuclear hormone receptor, glucocorticoid receptor (GR) to generate resistance. ARE-1 interferes with both AR- and GR-driven gene expression in enzalutamide-resistant LREX' cells, and is not rescued by GR activation. Compared with the use of enzalutamide alone, the co-treatment of ARE-1 and enzalutamide could reduce tumor growth by 80% without significant toxicity in castrated mice bearing LREX' tumors.

|
Download:
|
| Fig. 3. Chemical structures of ARE-1 and Oct1 polyamide. | |
{kind=link}
Inoue et al. found Oct1, a ligand-dependent AR-interacting partner, coordinated genome-wide AR signaling for prostate cancer growth [39]. They designed Py-Im polyamides to target the Oct1-binding sequence and investigated their gene inhibition effect and antitumor activity. The structure of Oct1 polyamide was shown in Fig. 3. The results of chromatin immunoprecipitation (ChIP) assay demonstrated Oct1 recruited to the ACSL3 enhancer, ACSL3 mRNA expression and ACSL3 protein levels were all reduced by Oct1 polyamide treatment, indicating Oct1 polyamide was capable of suppressing ACSL3 expression. Moreover, the treatment with Oct1 polyamide could significantly suppress castrationresistant prostate cancer growth in vitro and in vivo.
Previous studies suggested that Py-Im polyamides could interfere with RNA polymerase Ⅱ (pol Ⅱ) transcription elongation without evidence of DNA damage [37]. However, the mechanism of pol Ⅱ inhibition by Py-Im polyamides was unclear. Strathern et al. found sequence-specific Py-Im polyamides could sterically block an elongating polymerase at the targeted binding site, and the pol Ⅱ elongation-complex could not be rescued by transcription factor ⅡS. Through molecular modeling, they discovered that the highly conserved motif in pol Ⅱ (Arg1386 and His1387 of Rpb1) could serve as a sensor of DNA minor groove and could stall pol Ⅱ elongation via steric clash with minor groove binders [40].
Mutations at codons 12 (G12D) and 13 (G13D) are the most frequent mutations in metastatic colon cancer [41]. However, there have not been effective methods to target activated KRAS for clinical use. Nagase et al. developed a series of Py-Im polyamide indole-seco-CBI conjugates to target KRAS codon 13 and 12 mutations [42, 43]. First, they evaluated the specificity and alkylating activity of five different Py-Im polyamide-CBI conjugates possessing two β-alanine units in different positions. The high-resolution denaturing polyacrylamide gel electrophoresis (PAGE) and surface plasmon resonance (SPR) assay showed that conjugate 5 (ⅡPIβPI-γ-PIβPP-indole-seco-CBI) had high reactivity towards G13D, with alkylation occurring at the A of the sequence 5′-ACGTCACCA-3′ (site 2). Conjugate 6 (ⅡPIβPI-γ-PIPβP-indoleseco-CBI) (Fig. 4), in which one Py unit in conjugate 5 was replaced by a β unit, showed high selectivity while weak alkylating activity at site 2. Other three conjugates displayed inaccurate DNA alkylation, indicating the position of β/β pairing in Py-Im polyamides directly reflected their sequence-specific DNA alkylation. Then they designed a compound, IPβⅡPI-γ-PIβPP-indoleseco-CBI (KR12) (Fig. 4), to target KRAS codon 12 mutants (5′- WCGCCWWCA-3′). In cell experiments, it was found that KR12 was sufficient to inhibit mutant KRAS and caused specific-sequence DNA damage as well as cell apoptosis. In xenograft models, they observed that the tumor growth was significantly suppressed by weekly injections of KR12 compared to CBI and conjugate 6. Moreover, KR12 displayed low host toxicity in KRAS-mutated but not wild-type tumors.

|
Download:
|
| Fig. 4. Chemical structures of Py-Im polyamide-indole-seco-CBI conjugates binding to KRAS codon 13 (conjugate 6) and 12 (KR12) mutations respectively. | |
{kind=link}
Py-Im polyamides have frequently been supplemented with an epigenetically active molecule to increase their nuclear localization and to be used as sequence-specific gene modulators, alkylating agents, or fluorescent probes [44, 45]. In order to deliver Py-Im polyamides to mitochondria accurately, Sugiyama et al. conjugated a mitochondria-penetrating peptide with Py-Im polyamide and designed a new type of mitochondria-specific polyamide, termed MITO-PIP [46]. The structure and transformed localization preference of MITO-PIP were showed in Figs. 5a and b, respectively. The colocalization experiment confirmed that MITOPIPs could precisely localize into the mitochondria. The designed LSP-specific MITO-PIP could inhibit mitochondrial transcription factor A binding with the light-strand promoter (LSP), and trigger targeted transcriptional suppression. MITO-PIPs were the first DNA-based programmable functional ligands that were capable of inhibiting targeted ND6 in mitochondria.

|
Download:
|
| Fig. 5. (a) Chemical structure of MITO-PIP for mitochondrial DNA sequence recognition. (b) Schematic illustration of transformed localization preference of MITO-PIPs. Copied with permission [46]. Copyright 2017, American Chemical Society. | |
{kind=link}
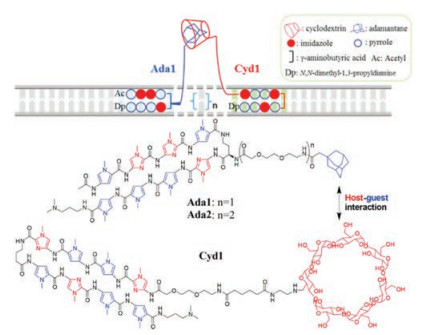
Sugiyama et al. also designed a new kind of Py-Im polyamides with a host-guest Cyd-Ada scaffold (Pip-HoGu) to mimic the cooperation of natural transcription factor pairs (Fig. 6) [47]. PIP1 and PIP2 were conjugated to Ada and Cyd respectively, affording PIP1-Ada (Ada1) and PIP2-Cyd (Cyd1). Using the DNA-binding sequences of cooperative Tax/CREB heterodimer as a module, Ada1 targets the Tax binding site (5′-WWGGCW-3′, W = A/T), host conjugate Cyd1 targets the CREB binding site (5′-WGWCGW-3′). The results of in vitro experiments (such as Tm, EMSA, and SPR assays) demonstrated Ada1 and Cyd1 were integrated to stable cooperative complexes with the ability of mimicking a TF pair. PipHoGu exhibited potently cooperative inhibitory effects on gene expression in cells, providing a promising strategy for precise manipulation of gene expression patterns.

|
Download:
|
| Fig. 6. Overview of a host-guest assemblies Ada1 and Cyd1 for mimicking transcription factor pairs. Copied with permission [47]. Copyright 2018, American Chemical Society. | |
{kind=link}
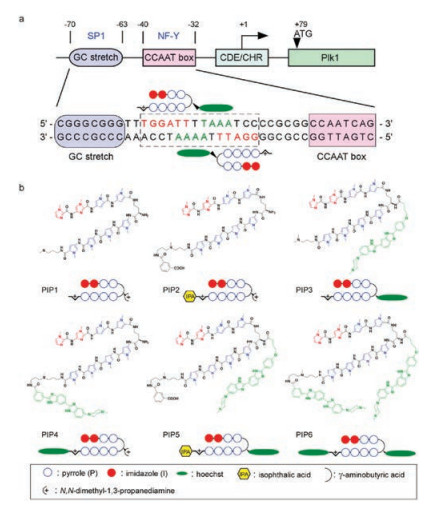
Focusing on developing a novel Polo-like kinase 1 (Plk1) inhibitor with high selectivity and efficacy, we designed and synthesized a series of Py-Im polyamide-hoechst conjugates to target the specific DNA sequence in the Plk1 promoter [48]. Initially, in searching for the potential targeted DNA sequence between two transcription factor binding sites, SP1 consensus site and NF-Y binding CCAAT box, we coincidently found a sequence 5′-GGATTTTAAATCC-3′, which possesses a pair of inverted repeats (5′-GGATTT-3′) and is highly A/T-rich (Fig. 7a). As we know, hoechst 33258 is a common nucleus-staining agent and binds with A/T-rich sequence in DNA. Therefore, conjugation of a hoechst 33258 derivative with the Py-Im polyamides was expected to improve the specificity to the targeted sequence by recognizing up to 10 bp. The structures of PIPs were shown in Fig. 7b. The results of immunoblotting assay showed that only PIP3, possessing a hoechst conjugation at the γ-turn position, had the ability to suppress Plk1 expression. The qRT-PCR results revealed that the Plk1 mRNA level was significantly reduced by the pre-treatment of cells with 10 nmol/L of PIP3 for 48 h. We next evaluated the in vivo antitumor activity of PIP3 against human lung cancer A549 xenograft, as well as non-small cell lung cancer model, NCI-H1299, and a breast cancer model, MDA-MB-231, indicating that PIP3 had great antitumor efficacy in a range of tumor types with low toxicity.

|
Download:
|
| Fig. 7. (a) Schematic representation of the binding of designed Py-Im polyamide to human Plk1 promoter. (b) Chemical structures of the synthetic Py-Im polyamide derivatives. Copied with permission [48]. Copyright 2017, American Association for Cancer Research. | |
{kind=link}
4.2. Effect of Py-Im polyamides at the chromatinized genome level
Py-Im polyamides not only could regulate single gene expression, but also direct the chromatin remodeling, which was mediated by histone acetylation and could induce global changes in the transcriptional status of a cell [49]. Small molecules that could inhibit histone deacetylase (HDAC), a chromatin-modifying enzyme, were proved to induce global changes in the acetylation profile but in a non-selective manner [5′]. Accordingly, conjugating these molecules with PIPs could render them distinctive recognition so that they were able to precisely modulate the complex transcriptional network. Sugiyama et al. have conjugated SAHA, a potent HDAC inhibitor, with selective Py-Im polyamides to induce endogenous pluripotency factors in mouse and germ cell genes in human fibroblasts, respectively [51, 52]. Through ChIP-seq studies, motif analysis and a specificity landscape study, they observed that different PIPs could direct SAHA to a set of silent genes and remodel the local chromosome, leading to different clusters of genes activation. To clarify this notion, they synthesized a library of 32 different SAHA-PIPs capable of recognizing distinct DNA sequence to evaluate their effect on the genome-wide gene expression [53]. They analyzed transcriptome profiles of HDFs cells treated with the 32 SAHA-PIPs by microarray separately, and revealed for the first time that each SAHA-PIP could activate a unique cluster of genes and noncoding RNAs. They also found that some SAHA-PIPs, rather than SAHA, had remarkable ability to activate therapeutically important genes like KSR2.
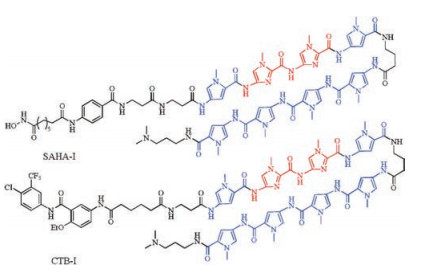
Considering the essential role of histone acetyltransferases (HATs) in gene regulation, they further conjugated an artificial HAT activator, N-(4-chloro-3-trifluoromethyl-phenyl)-2-ethoxy-benzamide (CTB) with a PIP (Ⅰ) to induce selective HAT-regulated genes. Through biological evaluation of CTB-Ⅰ using a microarray with appropriate controls (CTB, SAHA, and SAHA-Ⅰ), they found that CTB-Ⅰ and SAHA-Ⅰ (Fig. 8) activated about 26 and 40 genes, respectively, including 24 common genes, which indicated that CTB-Ⅰ activated a very similar cluster of genes to SAHA-Ⅰ. So they concluded that identical DNA-binding domain (Ⅰ) conjugated with distinct functional small molecules (HDAC inhibitor SAHA and HAT activator CTB) could have identical bioactivity [54].

|
Download:
|
| Fig. 8. Structures of SAHA-Ⅰ and CTB-Ⅰ for activating an identical gene network. | |
{kind=link}
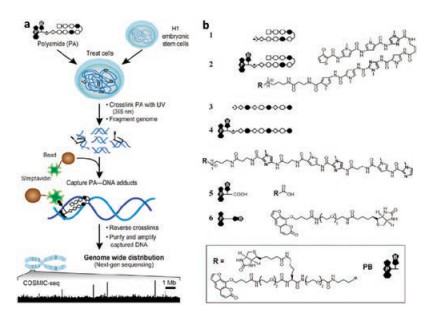
Ansari et al. designed trifunctional derivatives containing hairpin or linear PIPs, photo cross-linker psoralen and biotin to map genome-wide binding profiles of these two polyamides in living human cells by COSMIC-Seq [55]. The scheme of COSMICSeq and the structures of PIPs were showed in Figs. 9a and b, respectively. They found that the synthetic genome readers could bind to cognate sites across diverse chromatin states within the genome of live cells. Unexpectedly, they observed that PIPs could bind to repressive heterochromatin and actively transcribed euchromatin, which was different to natural TFs. The ability of PIPs to access heterochromatin maybe used to design chromatin remodeling machine, consisting of PIPs (DNA binding domain) and artificial activators (transcriptional activation domain). It may access to specific part of chromosome to regulate gene networks that direct cellular fate and function. The genome-directed design of molecules will be a significant strategy for precision-targeted therapeutics.

|
Download:
|
| Fig. 9. (a) The scheme illustration of COSMIC-seq. (b) The chemical structures of PyIm polyamide derivatives used in genome-wide sequencing. Copied with permission [55]. Copyright 2016, National Academy of Sciences. | |
{kind=link}
4.3. Recognition of telomeres by Py-Im polyamides
Telomeres are localized at each end of chromosome and play important roles in several life processes, such as chromosomes replication, aging and cancer. The number of telomere repeats in human cells decreases with cell division, relating to the aging process or tumorigenesis. Human telomeric DNA are consisting of the double-stranded tandem repeats 5′-TTAGGG-3′ and singlestranded 5′-TTAGGG-3′ overhangs, which tends to form G-quadruplexes or t-loops. These subchromosomal structures are ideal and interesting polyamide targets. One of the initial study reported by Laemmli et al. was that tandem hairpin Py-Im polyamides were able to target insect and vertebrate telomeres with excellent selectivity [56]. Afterwards, Sugiyama et al. developed Py-Im polyamides conjugated with alkylating agents to target telomeres and investigated their influence on the proliferation of cancer cells [57, 58]. To improve sequence specificity, they used a heterotrimeric mixture of Py-Im polyamides and distamycin A to target telomere repeats [59]. In recent years, they designed and synthesized a series of tandem hairpin Py-Im polyamides to recognize longer telomeres with higher specificity.
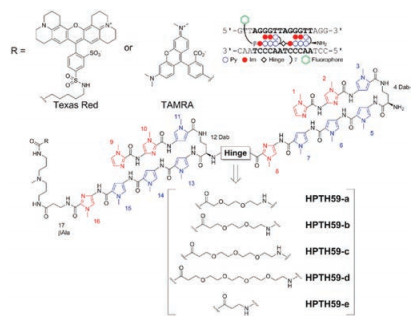
First, they prepared the tandem-hairpin motif of Py-Im polyamide-CBI conjugates with 12 bp recognition of telomeres, which could be used as a telomere alkylator with high selectivity [18]. Cell experiments demonstrated this conjugate could induce apoptotic cell death with lower cytotoxicity than single hairpin PyIm polyamide-CBI conjugates. Subsequently, they optimized the size of hinge region, which was important for matching the TTAGGG-seq repeat [60]. They synthesized several tandem hairpin PIPs with different-sized hinge segments (Fig. 10). The results of binding assay, human telomere staining and microheterogeneity demonstrated the optimal compound was TAMRA-labeled HPTH59-b with a hinge segment [-NH(C2H4O)2(C2H4)CO-]. Using HPTH59-b, they realized telomere visualization in mouse and human tissue sections [61]. It was proved that HPTH59-b could distinguish different telomere lengths between tumor and nontumor cells in tissues through quantitative measurement of telomere signals.

|
Download:
|
| Fig. 10. Synthesized tandem PIPs library with different-sized hinge segments. Copied with permission [60]. Copyright 2014, American Chemical Society. | |
{kind=link}
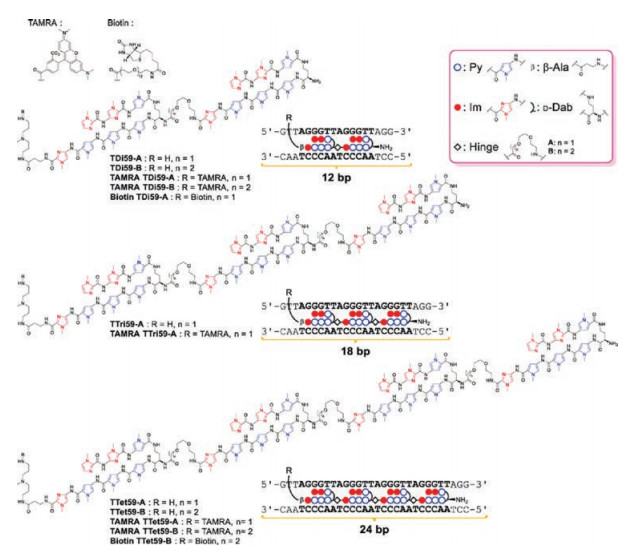
To increase the selectivity of tandem Py-Im polyamides to telomeres, they synthesized tandem trimers (Fig. 11), consisting of three hairpin subunits and two hinges, to targeted 18 bp in human telomeric repeats (TTAGGG)n [19]. Compared to previously mentioned tandem hairpin TAMRA TH59, the new tandem trimer TAMRA TT59 stained telomeres with lower background signal, showing higher selectivity to telomeres. The tandem tetramers (Fig. 11) were also synthesized to target 24 bp in human telomeres, which were composed of four hairpin units and three hinges [20]. Having much lower background signals, the tandem tetramers could be used to assay the binding of telomeres in the whole genome using high-throughput sequencing technology. To date, 24 bp is the longest binding site of synthetic, non-nucleic-acid based, sequence-specific DNA-binding molecules.

|
Download:
|
| Fig. 11. Chemical structures of tandem dimer, tandem trimer and tandem tetramer to target 12, 18 and 24 bp in human telomere sequence, respectively. Copied with permission [20]. Copyright 2016, American Chemical Society | |
{kind=link}
4.4. Py-Im polyamides used on nano-platform
Py-Im polyamides have been used to drive self-assembly on nano-platform based on the specific recognition of dsDNA via Watson-Crick base-pairing regimes. The first attempt to use Py-Im polyamides for construction of DNA nanoarchitectures was made by Heckel et al. [62, 63]. In their concept, Py-Im polyamides were called "DNA-strut", because they were recruiting and holding together two DNA helices and acted as a strong "glue" for DNA. They used dsDNA minicircles equipped with sequence-specific polyamides to build DNA catenanes (Fig. 12) [64]. The two DNA minicircles self-assembled into homodimeric complexes with interlocked C-shapes. Followed by the addition of a ring-closing oligonucleotide, DNA catenanes were created and would be useful components for making molecular motors.

|
Download:
|
| Fig. 12. Schematic representation of the catenane synthesis using DNA equipped with Py-Im polyamides. Copied with permission [64]. Copyright 2011, American Chemical Society. | |
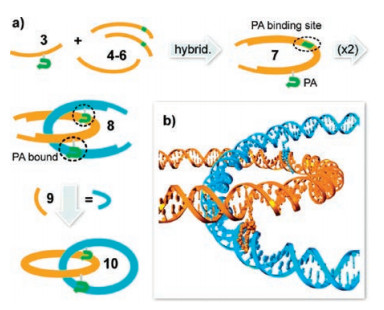{kind=link}
Graham et al. presented the first report that Py-Im polyamide functionalized AuNPs could direct assembly/disassembly of AuNPs [65]. They prepared PIP-functionalized AuNPs and DNA functionalized AuNPs. When matched ssDNA were added, these two AuNPs self-assembled to large particles by the specific binding of Py-Im polyamides with dsDNA, resulting in aggregation and colorimetric response (Fig. 13). This is a new concept that the assembly of AuNPs can be directed by the recognition capabilities of Py-Im polyamides to dsDNA selectively.

|
Download:
|
| Fig. 13. The assembly of AuNPs directed by Py-Im polyamides. Copied with permission [65]. Copyright 2012, American Chemical Society. | |
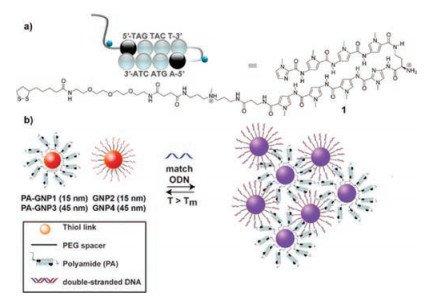{kind=link}
The role and function of Py-Im polyamides on nano-platform were also exhibited by an artificial nanoparticle-based transcription factor, termed NanoScript, designed by Lee et al. [66]. Transcription factors are master regulators of transcriptional activity and gene expression, containing three essential domains, which are nuclear localization signal (NLS) domain, DNA-binding domain (DBD) and activation domain (AD). In order to mimic the structure and function of TFs, NanoScript was constructed by tethering NLS peptide(NLS), Py-Im polyamides (DBD) and a transactivation peptide (AD) on a single AuNP to emulate the gene-regulating function of TFs (Fig. 14). Their results demonstrated NanoScript was able to mimic natural TFs and effectively initiate transcriptional activity of endogenous genes. They again designed NanoScript-MRF specific for myogenic regulatory factors (MRFs) to induce muscle cell differentiation [67]. They subsequently integrated CTB into NanoScript to enhance chondrogenesis of stem cells. They observed that NanoScript with both AD and CTB showed the greatest activity of histone acetyltransferase and highest expression of chondrogenic genes, possibly due to the cooperation of AD and CTB together on NanoScript [68]. As a proofof-concept experiment, NanoScript is an effective and tunable platform that can manipulate any targeted genes of interest, and can be utilized for other applications involving gene manipulations such as cancer therapy and cellular reprogramming.

|
Download:
|
| Fig. 14. Schematic representation of NanoScript mimicking transcription factors. Copied with permission [66]. Copyright 2014, American Chemical Society. | |
{kind=link}
5. Outlook
Py-Im polyamides, possessing higher affinity and specificity, as well as longer targeted sequence, have displayed powerful functions in regulation of gene expression and transcription, and have the potent to become a new class of gene-targeted therapeutics. Compared with other peptide or chemical drugs, PyIm polyamides are fully resistant to biological degradation by nucleases and do not induce unnecessary normal cell damage and carcinogenesis. It is more important that significantly deleterious effects in mice or rats have not been observed by intravenous or peritoneal injection of Py-Im polyamides [32, 69]. These advantages suggest that Py-Im polyamides are promising candidates for clinical applications. However, their long-term toxicity and stability are unclear, so further investigations about the effects of selected polyamides in preclinical animal models is crucial.
AcknowledgmentsThis work was supported by the Shenzhen Sciences & Technology Innovation Council (Nos. GJHS20170314154409805, GJHS20160331195142827, GJHS2015′417103343317, JCYJ201708 1815358196, JCYJ2015′40115′223649 and JCYJ20170413165 916608) and the National Natural Science Foundation of China (Nos. 21672254, 21778068, 215′2219 and 21432003).
| [1] |
L.H. Hurley, Nat. Rev. Cancer 2 (2002) 188-200. DOI:10.1038/nrc749 |
| [2] |
A. Ali, S. Bhattacharya, Bioorg. Med. Chem. 22 (2014) 4506-4521. |
| [3] |
P.B. Dervan, B.S. Edelson, Curr. Opin. Struct. Biol. 13 (2003) 284-299. DOI:10.1016/S0959-440X(03)00081-2 |
| [4] |
M.S. Blackledge, C. Melander, Bioorg. Med. Chem. 21 (2013) 6101-6114. DOI:10.1016/j.bmc.2013.04.023 |
| [5] |
P.B. Dervan, R.W. Burli, Curr. Opin. Chem. Biol. 3 (1999) 688-693. DOI:10.1016/S1367-5931(99)00027-7 |
| [6] |
C. Melander, R. Burnett, J.M. Gottesfeld, J. Biotechnol. 112 (2004) 195-220. DOI:10.1016/j.jbiotec.2004.03.018 |
| [7] |
P.B. Dervan, R.M. Doss, M.A. Marques, Curr. Med. Chem. Anti-Cancer Agents 5 (2005) 373-387. DOI:10.2174/1568011054222346 |
| [8] |
E.E. Baird, P.B. Dervan, J. Am. Chem. Soc. 118 (1996) 6141-6146. DOI:10.1021/ja960720z |
| [9] |
N.R. Wurtz, J.M. Turner, E.E. Baird, et al., Org. Lett. 3 (2001) 1201-1203. DOI:10.1021/ol0156796 |
| [10] |
J.M. Belitsky, D.H. Nguyen, N.R. Wurtz, et al., Bioorg. Med. Chem. 10 (2002) 2767-2774. DOI:10.1016/S0968-0896(02)00133-5 |
| [11] |
D.A. Harki, N. Satyamurthy, D.B. Stout, et al., Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 13′39-13′44. DOI:10.1073/pnas.0709492105 |
| [12] |
D.M. Chenoweth, D.A. Harki, P.B. Dervan, J. Am. Chem. Soc. 131 (2009) 7175-7181. DOI:10.1021/ja901307m |
| [13] |
J.W. Puckett, J.T. Green, P.B. Dervan, Org. Lett. 14 (2012) 2774-2777. DOI:10.1021/ol3010003 |
| [14] |
A.J. Fallows, I. Singh, R. Dondi, et al., Org. Lett. 16 (2014) 4654-4657. DOI:10.1021/ol502203y |
| [15] |
W. Su, S.J. Gray, R. Dondi, et al., Org. Lett. 11 (2009) 3910-3913. DOI:10.1021/ol9015139 |
| [16] |
L.J. Fang, G.Y. Yao, Z.Y. Pan, et al., Org. Lett. 17 (2015) 158-161. DOI:10.1021/ol503388a |
| [17] |
Y. Kawamoto, T. Bando, F. Kamada, et al., J. Am. Chem. Soc. 135 (2013) 16468-16477. DOI:10.1021/ja406737n |
| [18] |
M. Yamamoto, T. Bando, Y. Kawamoto, et al., Bioconjugate Chem. 25 (2014) 552-559. DOI:10.1021/bc400567m |
| [19] |
Y. Kawamoto, A. Sasaki, K. Hashiya, et al., Chem. Sci. 6 (2015) 2307-2312. DOI:10.1039/C4SC02339K |
| [20] |
Y. Kawamoto, A. Sasaki, A. Chandran, et al., J. Am. Chem. Soc. 138 (2016) 14100-14107. DOI:10.1021/jacs.6b09023 |
| [21] |
M. Mrksich, M.E. Parks, P.B. Dervan, J. Am. Chem. Soc. 116 (1994) 7983-7988. DOI:10.1021/ja00097a004 |
| [22] |
C.S. Jacobs, P.B. Dervan, J. Med. Chem. 52 (2009) 7380-7388. DOI:10.1021/jm900256f |
| [23] |
F. Yang, N.G. Nickols, B.C. Li, et al., J. Med. Chem. 56 (2013) 7449-7457. DOI:10.1021/jm401100s |
| [24] |
J.A. Raskatov, J.O. Szablowski, P.B. Dervan, J. Med. Chem. 57 (2014) 8471-8476. DOI:10.1021/jm500964c |
| [25] |
S. Nishijima, Shinohara K.-i., T. Bando, et al., Bioorg. Med. Chem. 18 (2010) 978-983. DOI:10.1016/j.bmc.2009.07.018 |
| [26] |
B.B. Liu, S. Wang, K. Aston, et al., Org. Biomol. Chem. 15 (2017) 9880-9888. DOI:10.1039/C7OB02513K |
| [27] |
S. Wang, K. Aston, K.J. Koeller, et al., Org. Biomol. Chem. 12 (2014) 7523-7536. DOI:10.1039/C4OB01456A |
| [28] |
J.L. Meier, A.S. Yu, I. Korf, et al., J. Am. Chem. Soc. 134 (2012) 17814-17822. DOI:10.1021/ja308888c |
| [29] |
N. Fukuda, T. Ueno, Y. Tahira, et al., J. Am. Soc. Nephrol. 17 (2006) 422-432. DOI:10.1681/ASN.2005060650 |
| [30] |
J.A. Raskatov, J.L. Meier, J.W. Puckett, et al., Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 1023-1028. DOI:10.1073/pnas.1118506109 |
| [31] |
N.G. Nickols, P.B. Dervan, Proc. Natl. Acad. Sci. U. S. A. 104 (2007) 10418-10423. DOI:10.1073/pnas.0704217104 |
| [32] |
X.F. Wang, H. Nagase, T. Watanabe, et al., Cancer Sci. 101 (2010) 759-766. DOI:10.1111/cas.2010.101.issue-3 |
| [33] |
J. Syed, G.N. Pandian, S. Sato, et al., Chem. Biol. 21 (2014) 1370-1380. DOI:10.1016/j.chembiol.2014.07.019 |
| [34] |
N.G. Nickols, J.O. Szablowski, A.E. Hargrove, et al., Mol. Cancer Ther. 12 (2013) 675-684. DOI:10.1158/1535-7163.MCT-12-1040 |
| [35] |
A. Tsunemi, T. Ueno, N. Fukuda, et al., J. Mol. Med. 92 (2014) 509-521. |
| [36] |
K. Hayatigolkhatmi, G. Padroni, W. Su, et al., Blood Cells Mol. Dis. 69 (2018) 119-122. DOI:10.1016/j.bcmd.2017.11.002 |
| [37] |
F. Yang, N.G. Nickols, B.C. Li, et al., Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 1863-1868. DOI:10.1073/pnas.1222035110 |
| [38] |
A.A. Kurmis, F. Yang, T.R. Welch, et al., Cancer Res. 77 (2017) 2207-2212. DOI:10.1158/0008-5472.CAN-16-2503 |
| [39] |
D. Obinata, K. Takayama, K. Fujiwara, et al., Oncogene 35 (2016) 6350-6358. |
| [40] |
L. Xu, W. Wang, D. Gotte, et al., Proc. Natl. Acad. Sci. U. S. A. 113 (2016) 12426-12431. DOI:10.1073/pnas.1612745113 |
| [41] |
J. Neumann, E. Zeindl-Eberhart, T. Kirchner, et al., Pathol. Res. Pract. 205 (2009) 858-862. DOI:10.1016/j.prp.2009.07.010 |
| [42] |
R.D. Taylor, S. Asamitsu, T. Takenaka, et al., Chem. Eur. J. 20 (2014) 1310-1317. DOI:10.1002/chem.v20.5 |
| [43] |
K. Hiraoka, T. Inoue, R.D. Taylor, et al., Nat. Commun. 6 (2015) 6706. DOI:10.1038/ncomms7706 |
| [44] |
W. Su, C.R. Bagshaw, G.A. Burley, Sci. Rep. 3 (2013) 1883. DOI:10.1038/srep01883 |
| [45] |
W. Su, M. Schuster, C.R. Bagshaw, et al., Angew. Chem. Int. Ed. 50 (2011) 2712-2715. |
| [46] |
T. Hidaka, G.N. Pandian, J. Taniguchi, et al., J. Am. Chem. Soc. 139 (2017) 8444-8447. DOI:10.1021/jacs.7b05230 |
| [47] |
Z.T. Yu, C.X. Guo, Y.L. Wei, et al., J. Am. Chem. Soc. 140 (2018) 2426-2429. |
| [48] |
K. Liu, L.J. Fang, H.Y. Sun, et al., Mol. Cancer Ther. 17 (2018) 988-1002. DOI:10.1158/1535-7163.MCT-17-0747 |
| [49] |
S.D. Taverna, H.T. Li, A.J. Ruthenburg, et al., Nat. Struct. Mol. Biol. 14 (2007) 1025-1040. DOI:10.1038/nsmb1338 |
| [50] |
G.N. Pandian, H. Sugiyama, Biotechnol. J. 7 (2012) 798-809. DOI:10.1002/biot.v7.6 |
| [51] |
G.N. Pandian, Y. Nakano, S. Sato, et al., Sci. Rep. 2 (2012) 544. DOI:10.1038/srep00544 |
| [52] |
L. Han, G.N. Pandian, S. Junetha, et al., Angew. Chem. Int. Ed. 52 (2013) 13410-13413. DOI:10.1002/anie.201306766 |
| [53] |
G.N. Pandian, J. Taniguchi, S. Junetha, et al., Sci. Rep. 4 (2014) 3843. |
| [54] |
L. Han, G.N. Pandian, A. Chandran, et al., Angew. Chem. Int. Ed. 54 (2015) 8700-8703. DOI:10.1002/anie.201503607 |
| [55] |
G.S. Erwin, M.P. Grieshop, D. Bhimsaria, et al., Proc. Natl. Acad. Sci. U. S. A. 113 (2016) E7418-E7427. DOI:10.1073/pnas.1604847113 |
| [56] |
K. Maeshima, S. Janssen, U.K. Laemmli, Embo J. 20 (2001) 3218-3228. DOI:10.1093/emboj/20.12.3218 |
| [57] |
S. Sasaki, T. Bando, M. Minoshima, et al., J. Am. Chem. Soc. 128 (2006) 12162-12168. DOI:10.1021/ja0626584 |
| [58] |
R. Takahashi, T. Bando, H. Sugiyama, Bioorg. Med. Chem. 11 (2003) 2503-2509. |
| [59] |
G. Kashiwazaki, T. Bando, K.I. Shinohara, et al., Bioorg. Med. Chem. 17 (2009) 1393-1397. DOI:10.1016/j.bmc.2008.12.019 |
| [60] |
A. Hirata, K. Nokihara, Y. Kawamoto, et al., J. Am. Chem. Soc. 136 (2014) 11546-11554. DOI:10.1021/ja506058e |
| [61] |
A. Sasaki, S. Ide, Y. Kawamoto, et al., Sci. Rep. 6 (2016) 29261. DOI:10.1038/srep29261 |
| [62] |
T.L. Schmidt, C.K. Nandi, G. Rasched, et al., Angew. Chem. Int. Ed. 46 (2007) 4382-4384. DOI:10.1002/(ISSN)1521-3773 |
| [63] |
T.L. Schmidt, A. Heckel, Small 5 (2009) 1517-1520. DOI:10.1002/smll.v5:13 |
| [64] |
T.L. Schmidt, A. Heckel, Nano Lett. 11 (2011) 1739-1742. DOI:10.1021/nl200303m |
| [65] |
Z. Krpetic, I. Singh, W. Su, et al., J. Am. Chem. Soc. 134 (2012) 8356-8359. DOI:10.1021/ja3014924 |
| [66] |
S. Patel, D. Jung, P.T. Yin, et al., ACS Nano 8 (2014) 8959-8967. DOI:10.1021/nn501589f |
| [67] |
S. Patel, P.T. Yin, H. Sugiyama, et al., ACS Nano 9 (2015) 6909-6917. DOI:10.1021/acsnano.5b00709 |
| [68] |
S. Patel, T. Pongkulapa, P.T. Yin, et al., J. Am. Chem. Soc. 137 (2015) 4598-4601. DOI:10.1021/ja511298n |
| [69] |
H. Matsuda, N. Fukuda, T. Ueno, et al., Kidney Int. 79 (2011) 46-56. DOI:10.1038/ki.2010.330 |