 2018, Vol. 29
2018, Vol. 29 



Protein–protein interactions (PPIs) mediate various biological processes and induce proximity changes of protein characteristics, including subcellular localization, enzymatic activity and binding properties [1]. PPIs can occur between either structured protein domains or relatively short peptide stretches. Of note, interfaces of PPIs often exhibit an average area of 800–2000 Å and result in rather flatter surfaces than small molecular ligands that bind to defined protein pockets [2, 3]. Small molecules have been proven can only target on those proteins that have a hydrophobic pocket, but cannot be applied in protein interfaces that feature with large shallow surfaces. Natural products are frequently used for this purpose, but the obtaining of the diversified compound collection is tedious and thus the selection of natural products with high specificity is limited. While peptide therapeutics do not suffer from these limitations of their target-recognition capabilities, their pharmaceutical functions are restricted by the poor in vivo stability towards proteolytic enzymes and the intrinsic inability to traverse cell membranes [4-6].
This wide range of protein targets thus were thought to be "undruggable" within a long duration until growing researchers focus on mimicking α-helix starting from native peptides found at the binding surfaces of two proteins [7-10]. α-Helices, also 3.613- helices, accounting for approximately 30%–40% of ordered protein, play a major role in binding interactions with many of significantly biological receptor proteins [11-13]. α-Helices were stabilized by intramolecular hydrogen bonds between the carbonyl oxygen atom and the amide proton at position i and i + 4 as well as salt bridges between residues (e.g., glutamic acid and lysine) aligned on that same face [14]. Inspired by these stabilizing effects, researchers gain great interest and develop various methods in generating α-helices and one of the most established strategies is peptide stapling.
Early examples of peptide stapling involved the lactam formation between Lys and Glu/Asp residues and the assembly of disulfide bonds between thiol groups in Cys analogues [15-17]. After that, all-hydrocarbon stapling strategy developed by Verdine et al. has been considered as one of the most promising therapeutics for undruggable targets, taking advantage of Grubbs catalysts to crosslink on resin, via ruthenium-catalyzed olefin metathesis, two unnatural amino acids bearing olefinic side chains at (i, i + 4) or (i, i + 7) positions [18]. Along with this all-hydrocarbon stapling chemistry, stabilization of helix is fulfilled by side-chain constraints using various alternative stapling strategies developed in the past 20 years [19].
The tutorial review by Lau et al. [19] has compared and evaluated the various side-chain macrocyclization techniques used for peptide stapling in terms of their synthetic accessibility, potential to address biological questions, and current stage of development. In this review, herein we mainly reported the latest advances of peptide stapling in the last three years and summarized the novel stapling strategies based on different synthon aligned on the side chain of peptides.
Utilizing different peptide modifications (e.g., hydrogen-bond surrogates, β-strands and β-sheets, turn-inducing amino acids), we can design reasonable α-helix mimics as inhibitors of PPIs other than peptide stapling, and the readers should be directed to other excellent reviews covering the progress in these fields [20].
2. Peptide stapling strategies 2.1. Ring-closing metathesis bearing olefinic side chainsAlkene ring-closing metathesis (RCM) was first employed as a peptide stapling reaction by Blackwell and Grubbs [21], and then Schafmeister et al. reported a variant of metathesis stapling using α, α-disubstituted amino acids bearing olefinic side-chains of different lengths and stereochemistry [18]. This approach contained two features for stabilization of α-helices: 1) the methylation of α-carbon atoms; 2) the introduction of a covalent side chain to side chain cross-link on resin (Fig. 1A). This all-hydrocarbon stapling chemistry has been successfully used to design various peptide inhibitors with improved proteolytic stability, membrane permeability, and biological activity [22-26]. One notable example is a hydrocarbon-stapled peptide antagonist, ALRN-6924, of the oncogenic protein MDM2 and its homolog MDMX that functionally inhibit the tumor suppressor protein p53, which is currently in phase 2 clinical trials for advanced solid tumors and lymphomas [27-29].

|
Download:
|
| Fig. 1. Ring-closing metathesis bearing olefinic side chains. | |
To mimic interacting leucine and isoleucine residues, Speltz et al. created new amino acids that incorporate a methyl group in the γ-position of the stapling amino acids S5 and successfully incorporated them into a sequence derived from steroid receptor coactivator 2 [30]. The incorporation of a γ-methyl group in the R or S configuration at the i position of an i, i-4 stapled peptide enabled the hydrocarbon staple to effectively mimic branched hydrophobic residues, of which the introduction of S-methyl group could result in higher helical content. And this design can also be applicable in the non-interacting stapled residues (Fig. 1B).
Other than the design of novel λR or λs as an atypical olefinic stapling amino acids, Wilson and coworkers [31, 32] removed the α-substituted methyl group which was previously used to avoid the intrinsic helix-destabilizing effect of D-configured amino acids and designed a mono-substituted amino alkenyl amino acid as an effective reagent for creation of a hydrocarbon constraint (Fig. 1C). This kind of amino acid has been proved to couple more effectively in conventional Fmoc-mediated peptide synthesis and the resultant constraint may remove steric clashes with the target when compared to the di-substituted amino acid [33].
It is worth noting that our group [34] recently described a new series of amino acids that not only contain the native side chain but also carry the alkenyl arms that are needed for the ring-closing stapling chemistry (Fig. 1D). Introducing these novel amino acids into the β-catenin-binding domain of Axin (469–482), we obtained a new category of stapled peptides with the retention of the native side chains, exhibiting high α-helicity, strong proteolytic stability and good cell permeability. This new side-chain-retention stapling method would expand the scope of the all-hydrocarbon stapled peptide strategy by retaining some important peripheral residues for PPIs or preserving key hydrophilic side chains for improving water solubility. Additionally, Oba et al. [35] introduced cyclic α, α-disubstituted α amino acids into arginine-rich peptides and provided an additional staple in the side chain (Fig. 1E). The designed peptides showed enhanced and prolonged cell-penetrating abilities, a high resistance to protease and conformationally stable helical structures.
Despite its broad utility, RCM generally gives rise to a mixture of E- and Z-olefin isomers that can hinder efforts for the large-scale production and isolation of such complex molecules. To address this issue, Mangold and Grubbs reported the assessment of various factors that promote catalyst-directed RCM and ethenolysis on a variety of peptide substrates by varying the olefin type, peptide sequence, and placement of the olefin in macrocycle formation [36]. These methods allow for control over olefin geometry in peptides, facilitating their isolation and characterization.
2.2. Sulfhydryl-based thiol-ene and thiol-yne hydrothiolationThe thiol-ene click reaction is attractive due to its specificity to olefins and facile transformation and has been developed as peptide stapling strategies using cysteine residues and alkene substituted side chains [37, 38]. Wang and Chou reported the discovery of a peptide stapling and macrocyclization method using thiol-ene reactions between two cysteine residues and an α, ω-diene in high yields [39]. This new approach could selectively modify cysteine residues in native, unprotected peptides with a variety of stapling modifications for helix stabilization or general macrocyclization (Fig. 2). Furthermore, this group reported the application of thiol-yne/thiol-ene reactions to synthesize monoand bi-cyclic stapled peptides and proteins [40].

|
Download:
|
| Fig. 2. A two-component thiol-ene coupling in unprotected peptides by Wang and Chou [39]. | |
Li et al. reported the in-tether modification of chiral-centerinduced helical (CIH) peptides via thiol-ene hydrothiolation on resin, yielding two separable peptide diastereomers with signifi- cantly different helicity, as supported by circular dichroism (CD) and NMR spectroscopy [41]. This strategy provided the chance for investigating the influence of solely conformational differences upon the biochemical/biophysical properties of peptides. Furthermore, this group constrained peptides into helical conformations via synergistic effects of dual in-tether chiral centers [42]. And a reversible and versatile on-tether modification of CIH peptides was next developed via the S-alkylation reaction [43]. Notably, the major alkylated product retained the helical structure of its precursor peptide, although a pair of peptide epimers could be afforded via on-tether S-alkylation. As an important addition to the above i, i + 4 in-tether chiral center induced helicity strategy (CIH strategy), Li et al. carried out a systematic study of the effects of CIH strategy on i, i + 7 system, from an in-tether sulfoxide chiral center to an in-tether hydrocarbon chiral center (Fig. 3) [44].

Last but not the least, the photo-induced intramolecular thiolyne macrocyclization was developed for rapid access to short stapled peptides with enhanced biophysical properties [45]. The resulting new stapling peptides were conformationally constrained into helices with satisfying functional group tolerance.
2.3. Thiol-arylation/alkylation via nucleophilic substitution (SN)Buchwald et al. discovered a facile transformation between perfluoro-aromatic molecules and a cysteine thiolate and this new approach enabled them to introduce rigid perfluoro-aromatic staples in unprotected peptides via selectively modifying cysteine residues [46]. And encouraged by a palladium-mediated method for cysteine arylation using biaryl-dialkyl phosphine-based complexes as arylating reagents [47], this group detailed a method for a divergent i, i + 4 and i, i + 7 stapling of unprotected peptides by crosslinking two cysteine residues with bis-palladium organometallic reagents (Fig. 4A), introducing a diverse array of aryl and biaryl linkers [48].

|
Download:
|
| Fig. 4. Thiol-arylation/alkylation based stapling strategies. | |
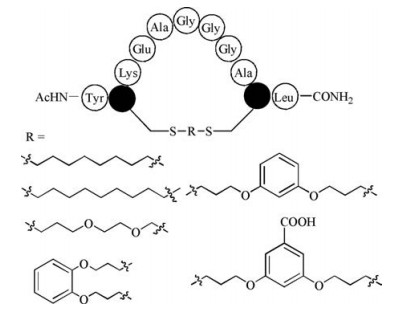
Different with the above palladium-mediated method, α, α'-dibromo-m-xylene (DBMB) was reported to stabilize synthetic peptides in α-helical conformations by Jo et al. in 2012 (Fig. 4B) [49]. They applied this strategy to the design of inhibitors of calpain that are based on calpastatin, and the resulting stapling peptides bear a two-turn α-helix that binds proximal to the active site cleft. Afterwards, other unsaturated halides including trans-1, 4- dibromo-2-butene (TDBB) and cis-1, 4-dichloro-2-butene (CDCB) have been applied as a thioether-bridged cross-link staple [50]. Compared to hydrocarbon stapled peptides, the ligands can be synthesized more easily, as no unnatural amino acids are required and the cyclization reaction is more efficient and yields no stereo isomers. Peraro et al. applied this strategy to produce autophagyinducing peptides that are intrinsically cell-penetrant aided by a diversity-oriented stapling approach. These stapling peptides bearing thioether-bridged cross-link could induce autophagy at micromolar concentrations in vitro and in vivo [51]. An atypical dibromomaleimide also was described to generate a cross-link with rapid and complete conversion [52]. This approach can be carried out on unprotected peptides using natural readily available amino acid side chains, extending the scope of stapling peptides aided by dibromomaleimide and related reagents (Fig. 4C). Another kind of heterocyclic stapling link, termed as s-tetrazine, has also been successfully introduced into unprotected peptides and the protein between two cysteine sulfhydryl groups by Brown and Smith [53], affording various macrocyclic peptides with a wide range of ring topology including i, i + 4, i, i + 7 and i, i + 11 (Fig. 4D). Of note, this kind of stapled conformations can be converted photochemically to their thiocyanate counterparts and subsequently the original peptide, thus comprising a readily removable peptide stapling, which is quite special from other stapling methods.
Other than thiol-based arylation, Lautrette et al. described a nitrogen arylation for the synthesis of macrocyclic peptide from unprotected precursors under efficient and mild conditions (Fig. 4E), which can be treated as a new stapling strategy [54]. The authors investigated various electrophiles and lysine-based nucleophiles, and the formation of N-C(aryl) bond could overcome the chemical stability issues sometimes encountered with cysteine S-arylation.
The formation of cross-link via other thiol-arylation/alkylation, although applied in macrocyclization chemistry now, can become the promising stapling strategy, such as the introduction of highly stable methylene thioacetal [55] and so on.
2.4. Azide-alkye cycloaddition based triazole linkageAs a biocompatible ligation technique, the Cu(Ⅰ)-catalysed azide alkyne cycloaddition (CuAAC) reaction is a clear candidate for peptide stapling, and Chorev et al. reported the first instance of affording stapling peptides at i, i + 4 position via azide-alkye cycloaddition [56].
Spring and coworkers developed an i, i + 7 double-click stapling technique, that is applied to inhibit the p53-MDM2 interaction [57, 58]. This two-component stapling strategy via double-click reaction using different functionalised dialkynyl linkers enables efficient synthetic access to a library of peptides bearing various staples. This approach enables a range of different stapled peptides to be efficiently generated by reacting a single linear diazido peptide with a collection of different dialkynyl stapling linkages (Fig. 5), including 1, 3-diethynylbenzene, linear and alternative aliphaticdiyne and so on [59-62].

|
Download:
|
| Fig. 5. Two-component stapling strategy via double-click reaction using differen functionalised dialkynyl linkers [59-62]. | |
This double-click stapling methodology has also been successfully applied in many PPIs for stabilizing α-helix such as p53- MDM2 [62], Ctf4 [61] and so on. Of note, Xu et al. reported the design of constrained peptide inhibitors having non-helical or extended conformations, targeting the tankyrase proteins (TNKS), poly(ADP-ribose) polymerases (PARP) that regulated Wnt signaling by targeting Axin for degradation [63]. And Wiedmann et al. also described a kind of cel permeable, non-helical constrained proteomimetics to target the HNF 1β-importin α PPI as the first constrained peptide nuclear import inhibitors [60]. In addition, Das et al. reported relevant application in designing epitope targeted macrocyclic peptide ligands via azide-alkye cycloaddition [64]. This kind of azide-alkye cycloaddition based triazole crosslink stapling strategy has been proven a promising solution for peptide therapeutics by extensive applications in various models.
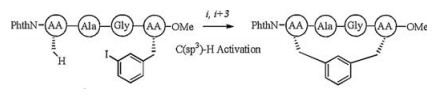
2.5. C-H activation based all-hydrocarbon linkageC-H activation emerged as an attractive method for the preparation of natural products [65], and soon was applied in peptide synthesis [66]. On the one hand, Mendive-Tapia et al. described a peptide stapling strategy through an intramolecular palladium-catalysed C(sp2)-H activation process, affording the unique constrained peptides featuring a covalent bond between tryptophan (Trp) and phenylalanine (Phe) or tyrosine (Tyr) residues [67]. Zhu et al. also accomplished the late-stage diversification of tryptophan-containing peptides through chemoand site-selective palladium-catalyzed C(sp2)-H arylation under exceedingly mild reaction conditions [68]. On the other hand, as a rarely addressed issue, the metal-catalyzed functionalization of C (sp3)-H bonds was also soon reported by Osberger et al. [69] and inspiringly applied in peptide stapling strategy. Noisier et al. developed a palladium-catalyzed late-stage C(sp3)-H activation method for peptide stapling, and prepared a library of unprecedented stapling peptides in solution at i, i + 3 and i, i + 4 position aided by a unique hydrocarbon cross-link (Fig. 6) [70].

|
Download:
|
| Fig. 6. C(sp3)-H activation based stapling strategy [70]. | |
Of note, in spite of an absence of typical stapling application, Tang et al. also described the development of a highly versatile peptide macrocyclization strategy through a palladium catalyzed C (sp3)-H activation and the synthesis of cyclic peptides featuring unique hydrocarbon linkages between the β-carbon of amino acids and the aromatic side chains of Phe and Trp [71].
2.6. Pre-installed stapling linkageAll of above stapling strategies begin from an assembly of a linear peptide whose side chain bears the stapling functional group, followed by a cyclization between the side chains to afford the corresponding stapling peptides. Actually, Liu and coworkers [72, 73] has examined the use of a diaminodiacid-based strategy for the solid-phase synthesis of peptide disulfide bond mimics with multiple cross-linking bridges, in which the desired disulfide surrogates could be readily pre-prepared in excellent yield, free of the limitation of the tedious and low-yielding folding process in Disulfide-rich peptides. Inspired by that, Li et al. [74] developed an alternative stapling strategy employing pre-installed diaminodiacid building blocks to introduce all-hydrocarbon staples into peptides by on-resin cyclization (Fig. 7). Avoiding the complicated cyclization reaction, a pre-installed stapling linkage can be introduced into the sequence under the condition of regular condensing agents. Of note, this new stapling method could provide an alternative way to obtain stapled peptides with tunable linkers of diaminodiacids more than i, i + 4 and i, i + 7.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3. Conclusion
Peptides offer great advantages over small molecular with potent specificity and high affinity, especially in inhibiting PPIs bearing swallow surfaces. However, two notable pharmacological weaknesses severely limit the widespread use of peptides as therapeutics: proteolytic stability and membrane permeability. In these regards, various elaborate side chain stapling strategies have been developed for α-helical peptides to circumvent these problems.
From the examples listed in this review, there is no denying that a single stapling strategy fails to address all cases, and the choice of stapling technique depends both on the biological questions being investigated and the individual nature of the PPI being studied. Although the most established all hydrocarbon cross-link stapling strategy is leading the way of this field, the development of nextgeneration alternative stapling techniques such as lactam and thioether cross-link, which combine the high membrane permeability of small molecules with the broad target ability of proteinbased drugs, will be the forefront of new drug discovery.
| [1] |
L.G. Milroy, T.N. Grossmann, S. Hennig, L. Brunsveld, C. Ottmann, Chem. Rev. 114 (2014) 4695-4748. DOI:10.1021/cr400698c |
| [2] |
N. London, B. Raveh, D. Movshovitz-Attias, O. Schueler-Furman, Proteins Struct. Funct. Bioinf. 78 (2010) 3140-3149. DOI:10.1002/prot.v78:15 |
| [3] |
I.S. Moreira, P.A. Fernandes, M.J. Ramos, Proteins Struct. Funct. Bioinf. 68 (2007) 803-812. DOI:10.1002/prot.21396 |
| [4] |
J.M. Mason, Future Med. Chem. 2 (2010) 1813-1822. DOI:10.4155/fmc.10.259 |
| [5] |
O. Keskin, A. Gursoy, B. Ma, R. Nussinov, Chem. Rev. 108 (2008) 1225-1244. DOI:10.1021/cr040409x |
| [6] |
V. Azzarito, K. Long, N.S. Murphy, A.J. Wilson, Nat. Chem. 5 (2013) 161-173. DOI:10.1038/nchem.1568 |
| [7] |
D.S. Nielsen, N.E. Shepherd, W. Xu, et al., Chem. Rev. 117 (2017) 8094-8128. DOI:10.1021/acs.chemrev.6b00838 |
| [8] |
M. Yang, K. Sunderland, C. Mao, Chem. Rev. 117 (2017) 10377-10402. DOI:10.1021/acs.chemrev.7b00100 |
| [9] |
K.R. Mahendran, A. Niitsu, L. Kong, et al., Nat. Chem. 9 (2017) 411-419. |
| [10] |
A.N. Zelikin, C. Ehrhardt, A.M. Healy, Nat. Chem. 8 (2016) 997-1007. DOI:10.1038/nchem.2629 |
| [11] |
H.M. Berman, J. Westbrook, Z. Feng, et al., Nucleic Acids Res. 28 (2000) 235-242. DOI:10.1093/nar/28.1.235 |
| [12] |
B.N. Bullock, A.L. Jochim, P.S. Arora, J. Am. Chem. Soc. 133 (2011) 14220-14223. DOI:10.1021/ja206074j |
| [13] |
O. Koch, J. Cole, P. Block, G. Klebe, J. Chem. Inf. Model. 49 (2009) 2388-2402. DOI:10.1021/ci900202d |
| [14] |
S. Marqusee, R.L. Baldwin, Proc. Natl. Acad. Sci. U. S. A. 84 (1987) 8898-8902. DOI:10.1073/pnas.84.24.8898 |
| [15] |
M. Chorev, E. Roubini, R.L. McKee, et al., Biochemistry 30 (1991) 5968-5974. DOI:10.1021/bi00238a022 |
| [16] |
D.Y. Jackson, D.S. King, J. Chmielewski, S. Singh, P.G. Schultz, J. Am. Chem. Soc. 113 (1991) 9391-9392. DOI:10.1021/ja00024a067 |
| [17] |
A.M. Felix, E.P. Heimer, C.T. Wang, et al., Int. J. Pept. Protein Res. 32 (1988) 441-454. |
| [18] |
C.E. Schafmeister, J. Po, G.L. Verdine, J. Am. Chem. Soc. 122 (2000) 5891-5892. DOI:10.1021/ja000563a |
| [19] |
Y.H. Lau, P. de Andrade, Y. Wu, D.R. Spring, Chem. Soc. Rev. 44 (2015) 91-102. DOI:10.1039/C4CS00246F |
| [20] |
C.J. White, A.K. Yudin, Nat. Chem. 3 (2011) 509-524. DOI:10.1038/nchem.1062 |
| [21] |
H.E. Blackwell, R.H. Grubbs, Angew. Chem. Int. Ed. 37 (1998) 3281-3284. DOI:10.1002/(ISSN)1521-3773 |
| [22] |
R.E. Moellering, M. Cornejo, T.N. Davis, et al., Nature 462 (2009) 182-188. DOI:10.1038/nature08543 |
| [23] |
L.D. Walensky, A.L. Kung, I. Escher, et al., Science 305 (2004) 1466-1470. DOI:10.1126/science.1099191 |
| [24] |
S. Baek, P.S. Kutchukian, G.L. Verdine, et al., J. Am. Chem. Soc. 134 (2012) 103-106. DOI:10.1021/ja2090367 |
| [25] |
F. Bernal, A.F. Tyler, S.J. Korsmeyer, L.D. Walensky, G.L. Verdine, J. Am. Chem. Soc. 129 (2007) 2456-2457. DOI:10.1021/ja0693587 |
| [26] |
P.S. Kutchukian, J.S. Yang, G.L. Verdine, E.I. Shakhnovich, J. Am. Chem. Soc. 131 (2009) 4622-4627. DOI:10.1021/ja805037p |
| [27] |
Y.S. Chang, B. Graves, V. Guerlavais, et al., Proc. Natl. Acad. Sci. U. S. A. 110 (2013) E3445-E3454. DOI:10.1073/pnas.1303002110 |
| [28] |
M. Wade, Y.C. Li, G.M. Wahl, Nat. Rev. Cancer 13 (2013) 83-96. DOI:10.1038/nrc3430 |
| [29] |
F. Meric-Bernstam, M.N. Saleh, J.R. Infante, et al., J. Clin. Oncol. 35 (2017) 2505. |
| [30] |
T.E. Speltz, S.W. Fanning, C.G. Mayne, et al., Angew. Chem. Int. Ed. 55 (2016) 4252-4255. DOI:10.1002/anie.201510557 |
| [31] |
J.A. Miles, D.J. Yeo, P. Rowell, et al., Chem. Sci. 7 (2016) 3694-3702. DOI:10.1039/C5SC04048E |
| [32] |
D.J. Yeo, S.L. Warriner, A.J. Wilson, Chem. Commun. 49 (2013) 9131-9133. DOI:10.1039/c3cc45231j |
| [33] |
C.H. Douse, S.J. Maas, J.C. Thomas, et al., ACS Chem. Biol. 9 (2014) 2204-2209. DOI:10.1021/cb500271c |
| [34] |
Y. Wu, Y.H. Li, X. Li, et al., Chem. Sci. 8 (2017) 7368-7373. DOI:10.1039/C7SC02420G |
| [35] |
M. Oba, M. Kunitake, T. Kato, A. Ueda, M. Tanaka, Bioconj. Chem. 28 (2017) 1801-1806. DOI:10.1021/acs.bioconjchem.7b00190 |
| [36] |
S.L. Mangold, R.H. Grubbs, Chem. Sci. 6 (2015) 4561-4569. DOI:10.1039/C5SC01507C |
| [37] |
C. Hoppmann, R. Kuhne, M. Beyermann, Beilstein J. Org. Chem. 8 (2012) 884-889. DOI:10.3762/bjoc.8.100 |
| [38] |
A.A. Aimetti, R.K. Shoemaker, C.C. Lin, K.S. Anseth, Chem. Commun. 46 (2010) 4061-4063. DOI:10.1039/c001375g |
| [39] |
Y. Wang, D.H.C. Chou, Angew. Chem. Int. Ed. 54 (2015) 10931-10934. DOI:10.1002/anie.201503975 |
| [40] |
Y. Wang, B.J. Bruno, S. Cornillie, et al., Chem. Eur. J. 23 (2017) 7087-7092. DOI:10.1002/chem.201700572 |
| [41] |
K. Hu, H. Geng, Q. Zhang, et al., Angew. Chem. Int. Ed. 55 (2016) 8013-8017. DOI:10.1002/anie.201602806 |
| [42] |
K. Hu, C. Sun, M. Yu, et al., Bioconj. Chem. 28 (2017) 1537-1543. DOI:10.1021/acs.bioconjchem.7b00171 |
| [43] |
K. Hu, C. Sun, Z. Li, Bioconj. Chem. 28 (2017) 2001-2007. DOI:10.1021/acs.bioconjchem.7b00321 |
| [44] |
K. Hu, C. Sun, D. Yang, et al., Chem. Commun. 53 (2017) 6728-6731. DOI:10.1039/C7CC03799F |
| [45] |
Y. Tian, J. Li, H. Zhao, et al., Chem. Sci. 7 (2016) 3325-3330. DOI:10.1039/C6SC00106H |
| [46] |
A.M. Spokoyny, Y. Zou, J.J. Ling, et al., J. Am. Chem. Soc. 135 (2013) 5946-5949. DOI:10.1021/ja400119t |
| [47] |
E.V. Vinogradova, C. Zhang, A.M. Spokoyny, B.L. Pentelute, S.L. Buchwald, Nature 526 (2015) 687-691. DOI:10.1038/nature15739 |
| [48] |
A.J. Rojas, C. Zhang, E.V. Vinogradova, et al., Chem. Sci. 8 (2017) 4257-4263. DOI:10.1039/C6SC05454D |
| [49] |
H. Jo, N. Meinhardt, Y. Wu, et al., J. Am. Chem. Soc. 134 (2012) 17704-17713. DOI:10.1021/ja307599z |
| [50] |
P. Diderich, D. Bertoldo, P. Dessen, et al., ACS Chem. Biol. 11 (2016) 1422-1427. DOI:10.1021/acschembio.5b00963 |
| [51] |
L. Peraro, Z. Zou, K.M. Makwana, et al., J. Am. Chem. Soc. 139 (2017) 7792-7802. DOI:10.1021/jacs.7b01698 |
| [52] |
C.M. Grison, G.M. Burslem, J.A. Miles, et al., Chem. Sci. 8 (2017) 5166-5171. DOI:10.1039/C7SC01342F |
| [53] |
S.P. Brown, A.B. Smith, J. Am. Chem. Soc. 137 (2015) 4034-4037. DOI:10.1021/ja512880g |
| [54] |
G. Lautrette, F. Touti, H.G. Lee, P. Dai, B.L. Pentelute, J. Am. Chem. Soc. 138 (2016) 8340-8343. DOI:10.1021/jacs.6b03757 |
| [55] |
C.M.B.K. Kourra, N. Cramer, Chem. Sci. 7 (2016) 7007-7012. DOI:10.1039/C6SC02285E |
| [56] |
M. Scrima, A. Le Chevalier-Isaad, P. Rovero, et al., Eur. J. Org. Chem. 2010 (2010) 446-457. DOI:10.1002/ejoc.v2010:3 |
| [57] |
Y.H. Lau, P. de Andrade, N. Skold, et al., Org. Biomol. Chem. 12 (2014) 4074-4077. DOI:10.1039/C4OB00742E |
| [58] |
Y.H. Lau, P. de Andrade, S.T. Quah, et al., Chem. Sci. 5 (2014) 1804-1809. DOI:10.1039/C4SC00045E |
| [59] |
Y.H. Lau, Y. Wu, P. de Andrade, W.R.J.D. Galloway, D.R. Spring, Nat. Protoc. 10 (2015) 585-594. DOI:10.1038/nprot.2015.033 |
| [60] |
M.M. Wiedmann, Y.S. Tan, Y. Wu, et al., Angew. Chem. Int. Ed. 56 (2017) 524-529. DOI:10.1002/anie.201609427 |
| [61] |
Y. Wu, F. Villa, J. Maman, et al., Angew. Chem. Int. Ed. 56 (2017) 12866-12872. DOI:10.1002/anie.201705611 |
| [62] |
Y.H. Lau, Y. Wu, M. Rossmann, et al., Angew. Chem. Int. Ed. 54 (2015) 15410-15413. DOI:10.1002/anie.201508416 |
| [63] |
W. Xu, Y.H. Lau, G. Fischer, et al., J. Am. Chem. Soc. 139 (2017) 2245-2256. DOI:10.1021/jacs.6b10234 |
| [64] |
S. Das, A. Nag, J. Liang, et al., Angew. Chem. Int. Ed. 54 (2015) 13219-13224. DOI:10.1002/anie.201505243 |
| [65] |
J. Yamaguchi, A.D. Yamaguchi, K. Itami, Angew. Chem. Int. Ed. 51 (2012) 8960-9009. DOI:10.1002/anie.201201666 |
| [66] |
A.F.M. Noisier, M.A. Brimble, Chem. Rev. 114 (2014) 8775-8806. DOI:10.1021/cr500200x |
| [67] |
L. Mendive-Tapia, S. Preciado, J. García, et al., Nat. Commun. 6 (2015) 7160. DOI:10.1038/ncomms8160 |
| [68] |
Y. Zhu, M. Bauer, L. Ackermann, Chem. Eur. J. 21 (2015) 9980-9983. DOI:10.1002/chem.201501831 |
| [69] |
T.J. Osberger, D.C. Rogness, J.T. Kohrt, A.F. Stepan, M.C. White, Nature 537 (2016) 214-219. DOI:10.1038/nature18941 |
| [70] |
A.F.M. Noisier, J. García, I.A. Ionuţ, F. Albericio, Angew. Chem. Int. Ed. 56 (2017) 314-318. DOI:10.1002/anie.201608648 |
| [71] |
J. Tang, Y. He, H. Chen, W. Sheng, H. Wang, Chem. Sci. 8 (2017) 4565-4570. DOI:10.1039/C6SC05530C |
| [72] |
H.K. Cui, Y. Guo, Y. He, et al., Angew. Chem. Int. Ed. 52 (2013) 9558-9562. DOI:10.1002/anie.v52.36 |
| [73] |
Y. Guo, D.M. Sun, F.L. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 14276-14281. DOI:10.1002/anie.201500699 |
| [74] |
F.L. Wang, Y. Guo, S.J. Li, et al., Org. Biomol. Chem. 13 (2015) 6286-6290. DOI:10.1039/C5OB00741K |