 2018, Vol. 29
2018, Vol. 29 



Contemporary drugs can be roughly classified into three broad categories: traditional small molecule drugs, large biologics and medium-sized peptides between the two. Since 1922, when insulin was first developed for the treatment of diabetes, peptides have become recognized as a critical drug class for areas where small molecules are ineffective. This middle-ground class has several advantages over the other categories. Compared to small molecule drugs, peptides have the potential for higher target specificity and potency, along with better safety profiles; and compared to large biologics, small peptides are cheaper to produce because of easier standardization and quality control [1].
Peptides are typically defined as small proteins of up to 50 amino acids. More than 7000 peptides have been identified so far from a wide variety of natural sources. They have evolved to play important and diverse roles in human physiology. They can provide a first line of defense against invading microorganisms, act as ligands of membrane receptors and ion channels, and serve as a means of intercellular communication in the form of hormones, neurotransmitters, and growth factors. The versatile biological activities of peptides, together with the fact that they generally have high specificity, potency, and safety, yet low toxicity, make them attractive candidates for development as therapeutics. More than 60 peptides are currently approved or in the process of final approval for the treatment of human disease, most commonly indicated for metabolic disease and oncology [2]. With more than 600 peptide molecules in clinical and preclinical development, the number of peptide therapeutics is expected to quickly expand in the near future. However, peptides have a distinct set of limitations compared to small-molecules, the most prominent of which is high susceptibility to acid/base hydrolysis and proteolytic degradation [2]. Because of this low stability and other issues like aggregation, there still remain many peptides with significant therapeutic potential that have not been successfully developed into clinical stage drugs despite many years of sustained effort. Therefore, optimizing peptide properties through engineering represents a major and important step for improving the therapeutic use [3].
Many different methods, including peptide conjugation, fusion, glycosylation, cyclization, and mutation have been pursued to improve therapeutic properties of peptides. These approaches have varying degrees of success in extending the circulatory halflives of peptides, improving their properties like proteolytic stability, solubility, absorption through biological membranes, and decreasing their aggregation propensity. In this review, we mainly focus on the comparison of the methods used for optimizing these properties.
2. Circulatory half-life extensionCirculatory half-life of a therapeutic peptide can be regulated by several different mechanisms, including immune system recognition, renal filtration, hepatic clearance, and receptor-mediated clearance [4, 5]. Among these mechanisms, renal filtration is the major route for peptide clearance. The molecular weight of peptide therapeutics is below the renal filtration cut-off of ~50 kDa; therefore, they can be rapidly excreted by the kidney [6]. Peptide modifications that enlarge their molecule sizes are expected to extend their circulatory half-lives by minimizing renal filtration and clearance. Currently, most methods used for increasing circulatory half-lives of therapeutic peptides, such as HSA (human serum albumin) conjugation, Fc (crystallizable fragment of immunoglobulin) conjugation, PEG (polyethylene glycol) conjugation, and XTEN (extended recombinant polypeptide) conjugation, are based on this concept [7].
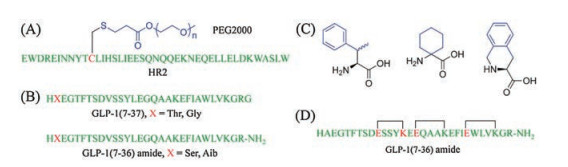
HSA conjugation is an important technique used to prolong circulatory half-life [8]. It can be achieved by directly linking a therapeutic peptide to HSA, which has a molecular weight of ~ 67 kDa and a serum half-life of 15–19 days in humans. The prolonged circulatory half-lives of peptide-HSA conjugates could be attributed to reduced glomerular filtration and the pHdependent interaction and recycling via the human neonatal Fc receptor (hFcRn) [9]. The first example of a peptide-HSA conjugate that is approved for marketing is GLP-1 (glucagon-like peptide-1)- HSA conjugate (Albigultide) (Fig. 1A). It was developed by Human Genome Sciences and GlaxoSmithKline and approved in 2014 for the treatment of type 2 diabetes. Through HSA conjugation, the circulatory half-life of GLP-1 was dramatically increased (from 2 min to 5 days) [10]. In addition to Albigultide, there are also many other peptide-HSA conjugates that are now either in development or in clinical trials [11].

|
Download:
|
| Fig. 1. Different methods that have been developed to improve circulatory half lives of therapeutic peptides. (A) HSA conjugation. (B) Fc conjugation. (C) PEG conjugation (D) XTEN conjugation. Peptide sequences are shown in green and mutated amino acids in red. Sar, Sarcosine. Nal, naphthyl alanine. | |
{kind=link}
Fc conjugation is another common method used to extend the circulatory half-life. Decreased clearance of peptide-Fc conjugate is mainly due to the pH-dependent binding of Fc to FcRn [12]. An example of a successful peptide-Fc conjugate is Dulaglutide, an injectable GLP-1 receptor agonist developed by Eli Lilly and approved in 2014. It was prepared by covalently linking GLP-1(7– 37) to the Fc of human immunoglobulin G4 (IgG4) (Fig. 1B). This conjugation extends the circulatory half-life of GLP-1 to approximately 5 days. In combination with other glucose-lowering drugs, Dulaglutide can be dosed once weekly to provide effective blood glucose control [13]. During the development of Dulaglutide and many other Fc conjugates, several important insights were revealed. First, the conjugation site (N- vs. C-terminus) and the length and structure of linker between peptide and Fc are essential for the activity of the resulting conjugate [14, 15]. Second, the Fc moiety should have minimal activity [7].
PEGylation is also well known to decrease the rate of glomerular filtration by the kidney. This is attributed to greatly increased molecular mass and hydrodynamic radius after being modified by large PEGs (>40 kDa) [16-18]. There are no PEGylated peptides currently used in clinic. Peginesatide, which was developed by Affymax and Takeda, was approved for the treatment of anemia in adult patients on dialysis in 2012 but was unfortunately withdrawn from the US market in 2014 due to serious hypersensitivity reactions by some patients (Fig. 1C). The reported mean halflife of peginesatide in dialysis patients was 47.9 ± 16.5 h following intravenous (Ⅳ) administration [19]. Previous studies proved that PEG size, architecture (linear, multi-arm or branched), as well as PEGylation site are all factors that influence circulatory half-lives of PEG conjugates [20, 21]. A drawback of PEGylation is the decreased biological activity of PEG-conjugates when compared to unmodified peptides. This is mainly due to the steric hindrance imposed by the PEG molecules, which would decrease the binding of peptides to their targets. Moreover, the covalent attachment of PEGs that are polydisperse in nature to therapeutic molecules often results in a population of conjugate species. Such conjugates may have different biological properties [16]. Although prolonged use of PEGylated drugs raised concerns of accumulation of non-biodegradable PEG in the liver and other organs [2], increased immunogenicity [22] and aggregation [23], to date, there are no reported safety issues that arose in connection with PEG at clinically relevant dose levels.
XTEN peptides, which were developed and optimized by Amunix in 2009 [24], are polymers that extend circulatory halflives in a manner similar to PEGs, but have the advantage of being homogeneous and biodegradable [25]. XTENs are made up of nonrepetitive sequences that are mainly composed of six natural amino acids, Ala, Glu, Gly, Pro, Ser, and Thr. Such sequences form the basis for their physicochemical properties, including the lack of secondary structure, high solubility and low aggregation propensity [24]. Since its development, XTENylation (also known as recombinant PEGylation, rPEG) has been widely tested as a replacement for PEGylation and many peptide-XTEN conjugates are in the discovery, preclinical, or clinical stages of development [7]. Many studies support the use of XTENylation to extend the half-lives of peptides. One such example is the development of VRS-859, an exenatide-XTEN conjugate that is still in phase Ⅰ trials by Diartis Pharmaceuticals for Type-2 diabetes mellitus [25]. This conjugate was obtained by fusion of an XTEN molecule to the Cterminus of exenatide (Fig. 1D). With this modification, the circulatory half-life of exenatide increased from 2.4 h to more than 5 days [25].
Besides the methods mentioned above, there are many other similar ways of extending circulatory half-lives of therapeutic peptides, including conjugation of peptides to macromolecules which have similar properties as albumin and Fc (transferrin), PEG (carbohydrate polymers) or XTEN (homoamino acid or Pro-Ala-Ser polymers), and covalent attachment of peptides to relatively small molecules like fatty acid and N- and O-linked glycans (Fig. 2) [2, 7, 26, 27]. The main reason behind increased circulatory half-life upon conjugation to macromolecules is thought to be similar to that mentioned above, although some small differences may exist [7]. For example, in addition to their large size, the presence of a large amount of negative charges on the surface of some carbohydrate polymers can further decrease the renal clearance of the resulting conjugates because of the electric force of repulsion between their surfaces and the negatively charged surfaces of glomerular capillaries [7]. The mechanisms of fatty acid and N- and O-glycan induced extension of circulatory half-life are different from those of macromolecules. Prolonged half-lives of fatty acid modified peptides are thought to result from the binding of fatty acid to albumin in human blood, whereas the lower clearance rates of glycosylated peptides are due to decreased receptor binding, internalization, and degradation.
One example of a successful drug that was developed using fatty acid conjugation is insulin detemir. It works as a long-acting insulin to improve glycemic control in adults with diabetes mellitus. Insulin detemir has a 14-C fatty acid, myristic acid, that is covalently attached to the lysine residue at position B29. The attached fatty acid increased the circulatory half-life of human insulin from ~ 4–6 min to 5–7 h (Fig. 2D) [28]. A drawback of fatty acid-based modification is that it reduces the water-solubility of peptides. Glycosylation of peptides has also been demonstrated to have a beneficial effect on the circulatory half-lives of many peptides, but have not yet been developed as therapeutic drugs. One reported example of using glycosylation to increase peptide circulatory half-life is the glycoengineering of human GLP-1. In this study, it was found that the attachment of sialylated Nacetyllactosamine to Asn residue was able to increase the circulatory half-life of human GLP-1 from a few minutes to a few hours (Fig. 2E) [29].

|
Download:
|
| Fig. 2. Additional methods that have been applied to improve circulatory half-lives of therapeutic peptides. (A) Transferrin conjugation. (B) Carbohydrate polymer conjugation (e.g., polysialylation). (C) PAS polymer conjugation (D) Fatty acid conjugation. (E) Glycosylation. | |
{kind=link}
3. Proteolytic stability increase
The utility of therapeutic peptides can also be hindered by rapid degradation in human blood and tissue due to the presence of many proteases and peptidases. These enzymes can inactivate peptides by splitting the molecules at specific internal peptide bonds, which reduces their bioavailability. To improve the application of peptides as therapeutics, many different methods have been utilized to increase their proteolytic resistance. These methods can be classified into three categories: (1) using large molecules (or macromolecules) to increase steric hindrance around cleavage sites; (2) changing amino acids in natural peptides to inhibit enzyme binding and action; and (3) increasing conformational rigidity of peptides to slow the rate of proteolytic hydrolysis. They could be used individually, or in combination to achieve the desired results.
Methods in the first category include peptide PEGylation, and conjugations to HSA or molecules with similar sizes. This can be illustrated by an example in which the half-life of a peptide was doubled after PEGylation (Fig. 3A) [30]. The engineered peptide was derived from the heptad repeat 2 (HR2) region of human immunodeficiency virus type 1 (HⅣ-1) glycoprotein 41 (gp41). It is able to prevent HⅣ-1 entry into cells but is susceptible to protease digestion. Its half-life in the presence of trypsin is 37 min. Upon attachment of a PEG moiety with molecular weight of ~2 kDa via the free cysteine at position 11, this half-life was increased to 91 min. This effect can be explained by the shielding effect of the PEG chains, which protects the HR2 peptide from attacks by trypsin. However, as explained above, this steric effect also led to a ~2-fold decrease in the inhibitory potency of the peptide.

|
Download:
|
| Fig. 3. Methods for increasing the proteolytic stability of peptides. (A) PEGylation. (B) Amino acid replacement. (C) Conformationally constrained amino acids. (D) Cyclization. | |
{kind=link}
Methods in the second category use proteinogenic and nonproteinogenic L-amino acids, D-amino acids, and backbone modified amino acids to replace the amino acids that are structural determinants for enzyme recognition and cleavage [31]. There are many examples of this type of research in literature. For example, it was found that substitution of Ala8 with Thr, Gly, Ser, and 2- aminoisobutyric acid (Aib) in GLP-1 enhanced its half-life in the presence of the enzyme dipeptidyl peptidase Ⅳ (DPP Ⅳ), which is responsible for the inactivation of GLP-1 (Fig. 3B). In this study, the half-life of GLP-1 (7–36) amide was determined to be 28 min, while the half-lives of the Gly8, Ser8, and Thr8 analogs were 159, 174, and 197 min, respectively. Aib8-GLP-1(7–36)-NH2 appeared to be completely resistant to DPP Ⅳ. As expected, mutations can affect peptide activity. Of these analogs, only Gly8 and Aib8 analogs were shown to have comparable binding affinity to natural GLP-1(7– 36)-NH2 [32, 33].
Methods in the third category improve the proteolytic stability of therapeutic peptides by increasing their structural rigidity. Increased rigidity could inhibit peptides from adopting optimal conformations for enzymatic cleavage, thus leading to enhanced stability. This can be achieved with peptide cyclization [34]. Peptides can be cyclized head-to-tail, side chain-to-side chain, side chain-to-end, or backbone-to-side chain through the formation of an amide bond or other chemically stable bonds such as lactone, ether, thioether, and disulfide bonds. The incorporation of amino acids that are capable of constraining a peptide conformation may also improve stability [35]. These amino acids either have sterically bulky side-chains or cyclized side chains (Fig. 3C). The impact of cyclization on proteolytic stability of peptides can be illustrated by a study of the cyclization of GLP-1. As shown in Fig. 3D, the introduction of three lactam bridges between residues 16 and 20, 22 and 26, 30 and 34 led to a significantly increased proteolytic resistance to both DPP Ⅳ (> 22 folds) and neutral endopeptidase (NEP) 24.11 (> 20 folds), and a slightly increased biological activity (> 2 folds) [36].
4. Solubility increase and aggregation decreaseLow solubility in aqueous solutions and/or a high propensity to aggregate into deposits has created a bottleneck in the development of peptides for clinical use. Many peptide drug candidates with great therapeutic potential are discarded simply because of unacceptable solubility and aggregation [37, 38]. As has been previously reported, the solubility and aggregation problems of peptides can be minimized or in some cases eliminated by altering amino acid sequences or conjugating peptides to highly soluble partners [39, 40].
One method for improving peptide solubility is to replace hydrophobic residues with hydrophilic or charged amino acids. It is suggested that this property arises from the water-binding ability of these amino acids [41] and their ability to strongly oppose close packing [42]. An example of such a modification is human calcitonin (hCT), in which several key residues involved in aggregation were replaced by hydrophilic and charged residues (Fig. 4A) [43]. These mutations led to a significant reduction in the rate of aggregation. The analysis showed that ~74% of the starting material for the mutated variant was still present in an aqueous buffer after incubation for 60 days, while hCT with an unmodified sequence had less than 1% remaining in the solution. More importantly, the new hCT variant was demonstrated to have increased receptor binding affinity and cAMP stimulation activity when compared to the natural hCT.

|
Download:
|
| Fig. 4. Methods for improving solubility and aggregation properties of peptides. (A) Mutation, (B) XTENylation, (C) Conjugation with low molecular weight solubility enhancement tags, (D) Glycosylation. | |
{kind=link}
The conjugation of therapeutic peptides to highly soluble partners has also been used to increase their solubility and/or decrease their aggregation rates. For example, XTEN was found to be effective as a solubilizing partner for human glucagon (hGCG), a highly efficacious drug for treating acute hypoglycemia [44]. Through conjugation of a 144 amino acid-long XTEN to its Cterminus, the solubility of hGCG was increased more than 60 folds (Fig. 4B) [44]. Similar to XTEN, PEG is also widely used in industry to improve the solubility of peptides [45]. Although XTENylation and PEGylation are effective methods for improving solubility and aggregation resistance, they have the drawback that the covalent attachment of large XTEN and PEG molecules to peptides often leads to changes in their biological activities.
Low molecular weight solubility enhancement tags [46, 47] and glycans can be used to overcome the drawback of large molecules. For example, it was reported that a betaine modification can reduce the aggregation propensity of the HⅣ entry inhibitor peptide CG-T20 and increase its solubility (~6.2 folds at pH 6), while not significantly affecting its biological functions (Fig. 4C) [47]. Similarly, a small O-linked glycan, a trimannose, was recently reported to significantly reduce insulin oligomerization without compromising its biological activity (Fig. 4D) [48].
5. Permeability enhancementPeptides typically have low cell membrane permeability, mainly due to their high flexibility, low lipophilicity, and high intermolecular hydrogen bonding capacity [49]. Therefore, many methods (e.g., cyclization, lipidization, N-methylation, and/or esterification of peptides) that increase peptide rigidity, reduce overall hydrophilicity, and/or reduce the number of amide hydrogen bond donors, were also found to confer membrane permeability [50-52]. This can be illustrated using the case of N-methylation of the Veber-Hirschmann cyclic hexapeptide, a highly active somatostatin agonist (Fig. 5A) [53]. An in vitro evaluation using a Caco-2 permeability model revealed that the permeability coefficient of an analog that was N-methylated at D-Trp8, Lys9, and Phe11 sites was significantly higher (68% increase) than that of the unmethylated parent form. The underlying reason for this increase was not determined, although it may be due to the reduced overall hydrogen bonding potential and increased overall lipophilicity [50].

|
Download:
|
| Fig. 5. Methods developed for improving the cell membrane permeability of peptides. (A) N-methylation. (B) Conjugation with cell-penetrating peptides. (C) Conjugation with simple nutrient molecules. | |
{kind=link}
Conjugation of peptides to cell-penetrating peptides (CPPs) can also lead to improved cell membrane permeability [54]. CPPs are short peptides that are capable of traversing the plasma membranes of cells. On the basis of their physical and chemical properties, they can be classified as cationic, amphipathic and hydrophobic CPPs. It is generally accepted that the principal internalization route for most CPPs and peptide-CPP conjugates is endocytosis [55]. CPP-conjugation has been used to increase the cell permeability of human parathyroid hormone (PTH) (1–34), a peptide that is important for regulating calcium and phosphorus metabolism. The engineering was carried out by attaching a nonaarginine (R9) peptide to the N-terminus of PTH (1–34) (Fig. 5B). The effect on permeability was evaluated using the Caco-2 permeability model. The results demonstrated that an N-terminal conjugation of R9 increased the permeability to approximately 3 folds as compared to that of the unmodified molecule [56].
Conjugation to small molecules can serve as another way of improving peptide permeability. Such small molecules are generally simple nutrient molecules that include vitamin B12, folic acid, and sugar, all of which increase the permeability of peptides through natural transport mechanisms [57, 58]. This has been demonstrated in the case of endomorphin-1, a peptide that is equipotent with morphine in producing analgesia. It was found that conjugation of lactose to the N-terminus of endomorphin-1 led to a significant increase (>700 folds) in its permeability across Caco-2 cell monolayer (Fig. 5C) [59].
6. Conclusions and outlookPeptides represent a great opportunity for the development of new drugs to address unmet medical needs. However, their poor physicochemical properties hamper their clinical application. Significant progress has been made over the last 40 years to overcome the problems associated with therapeutic peptides. A variety of different methods have been reported for improving the stability, solubility, permeability, and aggregation properties of peptides. These methods had varying degrees of success when applied to different peptides. Currently, there is no single method that has been deemed the most effective for peptide engineering, or proven to have beneficial effects on all properties of peptides. The very best peptide analogs might consist of several different modifications that individually give good results or have unforeseen synergies with each other. To exhaustively test all possible peptide engineering methods and their combinations, an enormous number of peptide variants would need to be synthesized and characterized. This would require an unreasonable expenditure of time and effort. Therefore, in the future, it would be advantageous to use the trove of empirical data gathered from previous studies to predict in silico, before synthesis, particular modifications or combinations of modifications that are likely to improve the therapeutic potential of peptides.
AcknowledgmentsWe are thankful for the support from the University of Colorado Boulder (Start-up Fund) and the National Science Foundation CAREER Award (No. CHE-1454925) during the course of this study.
| [1] |
J.L. Lau, M.K. Dunn, Bioorg. Med. Chem. 26 (2018) 2700-2707. DOI:10.1016/j.bmc.2017.06.052 |
| [2] |
L. Di, AAPS J. 17 (2015) 134-143. DOI:10.1208/s12248-014-9687-3 |
| [3] |
K. Fosgerau, T. Hoffmann, Drug Discov. Today 20 (2015) 122-128. DOI:10.1016/j.drudis.2014.10.003 |
| [4] |
L. Diao, B. Meibohm, Clin. Pharmacokinet. 52 (2013) 855-868. DOI:10.1007/s40262-013-0079-0 |
| [5] |
A. Muheem, F. Shakeel, M.A. Jahangir, et al., Saudi Pharm. J. 24 (2016) 413-428. DOI:10.1016/j.jsps.2014.06.004 |
| [6] |
R.E. Kontermann, Expert Opin. Biol. Ther. 16 (2016) 903-915. DOI:10.1517/14712598.2016.1165661 |
| [7] |
W.R. Strohl, BioDrugs 29 (2015) 215-239. DOI:10.1007/s40259-015-0133-6 |
| [8] |
P. Yeh, D. Landais, M. Lemaitre, et al., Proc. Natl. Acad. Sci. U. S. A. 89 (1992) 1904-1908. DOI:10.1073/pnas.89.5.1904 |
| [9] |
M.M. Schmidt, S.A. Townson, A.J. Andreucci, et al., Structure 21 (2013) 1966-1978. DOI:10.1016/j.str.2013.08.022 |
| [10] |
S. Madsbad, U. Kielgast, M. Asmar, etal., Diabetes Obes. Metab. 13 (2011) 394-407. |
| [11] |
M.T. Larsen, M. Kuhlmann, M.L. Hvam, K.A. Howard, Mol. Cell. Ther. 4 (2016) 3. DOI:10.1186/s40591-016-0048-8 |
| [12] |
J. Caravella, A. Lugovskoy, Curr. Opin. Chem. Biol. 14 (2010) 520-528. DOI:10.1016/j.cbpa.2010.06.175 |
| [13] |
C.A. Tibble, T.S. Cavaiola, R.R. Henry, Expert Rev. Endocrinol. Metab. 8 (2013) 247-259. DOI:10.1586/eem.13.20 |
| [14] |
G. Shimamoto, C. Gegg, T. Boone, C. Queva, MAbs 4 (2012) 586-591. DOI:10.4161/mabs.21024 |
| [15] |
W. Glaesner, A.M. Vick, R. Millican, et al., Diabetes Metab. Res. Rev. 26 (2010) 287-296. DOI:10.1002/dmrr.v26:4 |
| [16] |
F.M. Veronese, G. Pasut, Drug Discov. Today 10 (2005) 1451-1458. DOI:10.1016/S1359-6446(05)03575-0 |
| [17] |
F.M. Veronese, J.M. Harris, Adv. Drug Deliv. Rev. 54 (2002) 453-456. DOI:10.1016/S0169-409X(02)00020-0 |
| [18] |
M. Swierczewska, K.C. Lee, S. Lee, Expert Opin. Emerg. Drugs 20 (2015) 531-536. DOI:10.1517/14728214.2015.1113254 |
| [19] |
T. Hermanson, C.L. Bennett, I.C. Macdougall, Expert Opin. Drug Saf. 15 (2016) 1421-1426. DOI:10.1080/14740338.2016.1218467 |
| [20] |
Y.S. Youn, S.Y. Chae, S. Lee, et al., Biochem. Pharmacol. 73 (2007) 84-93. DOI:10.1016/j.bcp.2006.09.013 |
| [21] |
M. Weert, E.H. Møller, Immunogenicity of Biopharmaceuticals[M]. 1st ed.. New York: Springer-Verlag, 2008.
|
| [22] |
T. Ishida, M. Ichihara, X. Wang, H. Kiwada, J. Control. Release 115 (2006) 243-250. DOI:10.1016/j.jconrel.2006.08.001 |
| [23] |
D.S. Pisal, M.P. Kosloski, S.V. Balu-Iyer, J. Pharm. Sci. 99 (2010) 2557-2575. DOI:10.1002/jps.22054 |
| [24] |
V. Schellenberger, C.W. Wang, N.C. Geething, et al., Nat. Biotechnol. 27 (2009) 1186-1190. DOI:10.1038/nbt.1588 |
| [25] |
V.N. Podust, S. Balan, B.C. Sim, et al., J. Control. Release 240 (2016) 52-66. DOI:10.1016/j.jconrel.2015.10.038 |
| [26] |
S.B. van Witteloostuijn, S.L. Pedersen, K.J. Jensen, ChemMedChem 11 (2016) 2474-2495. DOI:10.1002/cmdc.v11.22 |
| [27] |
S.V. Moradi, W.M. Hussein, P. Varamini, P. Simerska, I. Toth, Chem. Sci. 7 (2016) 2492-2500. DOI:10.1039/C5SC04392A |
| [28] |
D.P. Begg, A.A. May, J.D. Mul, et al., Diabetes 64 (2015) 2457-2466. DOI:10.2337/db14-1364 |
| [29] |
T. Ueda, K. Tomita, Y. Notsu, et al., J. Am. Chem. Soc. 131 (2009) 6237-6245. DOI:10.1021/ja900261g |
| [30] |
M. Danial, T.H. van Dulmen, J. Aleksandrowicz, A.J. Potgens, H.A. Klok, Bioconjug. Chem. 23 (2012) 1648-1660. DOI:10.1021/bc3002248 |
| [31] |
D. Mathur, S. Prakash, P. Anand, et al., Sci. Rep. 6 (2016) 36617. DOI:10.1038/srep36617 |
| [32] |
B. Manandhar, J.M. Ahn, J. Med. Chem. 58 (2015) 1020-1037. DOI:10.1021/jm500810s |
| [33] |
C.F. Deacon, L.B. Knudsen, K. Madsen, et al., Diabetologia 41 (1998) 271-278. DOI:10.1007/s001250050903 |
| [34] |
S.H. Joo, Biomol. Ther. (Seoul) 20 (2012) 19-26. DOI:10.4062/biomolther.2012.20.1.019 |
| [35] |
O. Van der Poorten, A. Knuhtsen, D. Sejer Pedersen, S. Ballet, D. Tourwe, J. Med. Chem. 59 (2016) 10865-10890. DOI:10.1021/acs.jmedchem.6b01029 |
| [36] |
E.N. Murage, G. Gao, A. Bisello, J.M. Ahn, J. Med. Chem. 53 (2010) 6412-6420. DOI:10.1021/jm100602m |
| [37] |
L. Malavolta, M.R. Pinto, J.H. Cuvero, C.R. Nakaie, Protein Sci. 15 (2006) 1476-1488. DOI:10.1110/(ISSN)1469-896X |
| [38] |
P. Vlieghe, V. Lisowski, J. Martinez, M. Khrestchatisky, Drug Discov. Today 15 (2010) 40-56. DOI:10.1016/j.drudis.2009.10.009 |
| [39] |
A. Bak, D. Leung, S.E. Barrett, et al., AAPS J. 17 (2015) 144-155. DOI:10.1208/s12248-014-9688-2 |
| [40] |
S.R. Trevino, J.M. Scholtz, C.N. Pace, J. Pharm. Sci. 97 (2008) 4155-4166. DOI:10.1002/jps.21327 |
| [41] |
S.R. Trevino, J.M. Scholtz, C.N. Pace, J. Mol. Biol. 366 (2007) 449-460. DOI:10.1016/j.jmb.2006.10.026 |
| [42] |
I. Pallares, S. Ventura, Proteomics 16 (2016) 2570-2581. DOI:10.1002/pmic.v16.19 |
| [43] |
S.B. Fowler, S. Poon, R. Muff, et al., Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 10105-10110. DOI:10.1073/pnas.0501215102 |
| [44] |
N.C. Geething, W. To, B.J. Spink, et al., PLoS One 5 (2010) e10175. DOI:10.1371/journal.pone.0010175 |
| [45] |
I.W. Hamley, conjugates PEG-peptide, Biomacromolecules 15 (2014) 1543-1559. DOI:10.1021/bm500246w |
| [46] |
A. Kato, K. Maki, T. Ebina, et al., Biopolymers 85 (2007) 12-18. DOI:10.1002/bip.v85:1 |
| [47] |
J. Xiao, A. Burn, T.J. Tolbert, Bioconjug. Chem. 19 (2008) 1113-1118. DOI:10.1021/bc800063k |
| [48] |
X. Guan, P.K. Chaffey, X. Wei, et al., ACS Chem. Biol. 13 (2018) 73-81. DOI:10.1021/acschembio.7b00794 |
| [49] |
T. Rezai, J.E. Bock, M.V. Zhou, et al., J. Am. Chem. Soc. 128 (2006) 14073-14080. DOI:10.1021/ja063076p |
| [50] |
D.J. Craik, D.P. Fairlie, S. Liras, D. Price, Chem. Biol. Drug Des. 81 (2013) 136-147. DOI:10.1111/cbdd.2012.81.issue-1 |
| [51] |
A.H. Kahns, A. Buur, H. Bundgaard, Pharm. Res. 10 (1993) 68-74. DOI:10.1023/A:1018973029651 |
| [52] |
O. Zupancic, A. Bernkop-Schnurch, J. Control. Release 255 (2017) 242-257. DOI:10.1016/j.jconrel.2017.04.038 |
| [53] |
E. Biron, J. Chatterjee, O. Ovadia, et al., Angew. Chem. Int. Ed. 47 (2008) 2595-2599. DOI:10.1002/(ISSN)1521-3773 |
| [54] |
G. Guidotti, L. Brambilla, D. Rossi, Trends Pharmacol. Sci. 38 (2017) 406-424. DOI:10.1016/j.tips.2017.01.003 |
| [55] |
J.P. Richard, K. Melikov, E. Vives, et al., J. Biol. Chem. 278 (2003) 585-590. DOI:10.1074/jbc.M209548200 |
| [56] |
M. Kristensen, A.M. de Groot, J. Berthelsen, et al., Bioconjug. Chem. 26 (2015) 477-488. DOI:10.1021/bc5005763 |
| [57] |
S.T. Buckley, F. Hubalek, U.L. Rahbek, Tissue Barriers 4 (2016) e1156805. DOI:10.1080/21688370.2016.1156805 |
| [58] |
A.K. Petrus, T.J. Fairchild, R.P. Doyle, Angew. Chem. Int. Ed. 48 (2009) 1022-1028. DOI:10.1002/anie.v48:6 |
| [59] |
P. Varamini, F.M. Mansfeld, J.T. Blanchfield, et al., J. Med. Chem. 55 (2012) 5859-5867. DOI:10.1021/jm300418d |