 2018, Vol. 29
2018, Vol. 29 



b School of Science, Xihua University, Chengdu 610039, China
Protein-protein interaction (PPI) is the interaction between two proteins, either identical or different, using part of their domains to form interfaces, to regulate the function of the partners. PPIs are involved in most cellular processes in all levels of biological functions. It has been estimated that a total between 130000 and 600000 PPIs exists in the human interactome. Dysregulation of PPIs is associated with broad range of human diseases, many of which we do not have satisfactory therapies currently [1-5].
Both the academia and the pharmaceutical industry have devoted huge efforts in recent decades in this area, and tremendous progress has been made, with success of monoclonal antibodies and some small molecule drugs targeting PPIs. Antibody drugs have been successfully developed against a variety of targets and are approved for various human diseases, such as TNFα in autoimmune diseases, PCSK9 for hypercholestrolemia, PD- 1 and PD-L1 in different types of cancer, just to name a few. Venetoclax, a BCL-2 inhibitor with traditional drug-like structure, was approved by FDA in 2016 for chronic lymphocytic leukemia (CLL) associated with 17-p depletion. The drug has a MW of 868 and is orally administered, and is the first FDA approval of a protein-protein interaction inhibitor of BCL-2 [6].
However, compared to the huge number of PPIs and their potential in therapy, the ones already employed in therapeutic use are only a very tiny portion. High-throughput screening against PPI targets using small molecule compound libraries has not been as successful as it worked for other targets like enzymes. This is mainly because of the structural characteristics of PPIs. The interfaces of PPIs are usually flat, with an average area of 800–2000 Å2, and often without obvious pocket, cleft or groove. Therefore the screening often gives low hit rate [4, 5].
On the other hand, antibodies and other protein drugs are suitable for binding to the large, flat interface of PPI targets, and that is the reason for their high specificity, high efficiency, low toxicity and few side effects. However, because antibodies cannot cross the cell membrane, they have only been used against extracellular targets. For the vast number of intracellular PPI targets, new drug modalities are needed. For this purpose, peptides are good candidates. With molecular weight between small molecules and large proteins, the size of peptide is suitable for PPI interface binding. Peptides are in general biologically compatible, with low toxicity, and can achieve high selectivity. Because of these advantages, peptides and peptidomimetics are also widely used as probes in bioanalytical and biomedical research besides pharmaceutical development. However, native peptides often suffer with major drawbacks such as moderate potency, low protease stability, and usually are not permeable to cell membrane. To improve pharmacological properties of peptides, various chemical modifications have been developed, including L to D amino acid substitution, N-methylation, incorporation of turn mimetics, helix mimetics or other non-proteinogenic amino acids, using N-cap for α-helix nucleation, and cyclization.Among these methods, macrocyclization may provide peptides with improved target affinity through pre-organization, and usually increases the protease stability [7-18].
A diversity of chemistry has been developed for peptide macrocyclization, such as the formation of lactam, lactone, disulfide bond, thioether from Michael addition or nucleophilic substitution, olefin metathesis, and Huisgen 1, 3-dipolar cycloaddition. These reactions can be performed on-resin, thus incorporated into the solid-phase peptide synthesis conveniently [3-5, 7-10, 19]. An approach using pre-prepared orthogonally protected diaminodiacid building blocks, developed by Liu and collaborators, can also be used for the synthesis of macrocyclic peptides. Both allhydrocarbon and linkers containing heteroatoms or functional groups can be incorporated prior to solid-phase peptide synthesis, enabling straight forward synthesis of macrocyclic peptide with disulfide surrogates among others [20-22]. Reactions that are biocompatible, such as thioether formation and Huisgen 1, 3- dipolar cycloaddition, can be applied for peptide macrocyclization in biological systems. Macrocyclization of unprotected peptides can also be carried out under enzyme catalysis, e.g., by sortase A, provided that proper enzyme-recognizing sequence is incorporated [23, 24].
In general, there are two ways to discover macrocyclic peptides for PPI modulation, high-throughput screening including combinatorial chemistry and biological display technologies, and structure-based rational design. There are plenty of examples of hit identification of cyclic peptides for both approaches, and a combination of these two methods in the iterative optimization process following the hit identification is often employed [5, 8, 11-13].
2. Hit identification by high-throughput screeningCombinatorial peptide library synthesis and screening methods are now more often used in peptide lead optimization. Various display methods using biological systems like phage, bacteria, yeast, and in vitro protein expression, are used in initial peptide hit generation instead. A combination of chemistry and biology methods has been used to adapt these technologies for macrocyclic peptide library generation and screening. Following are some prominent examples.
2.1. Phage displayBacteriophage is engineered to uniformly display a library of peptides on the surface. The library is applied to the immobilized target protein, and those phages having peptide sequence with affinity to the target may stay on the target and those without affinity are washed away. After several rounds of panning, the sequences with highest affinity may be isolated [25].
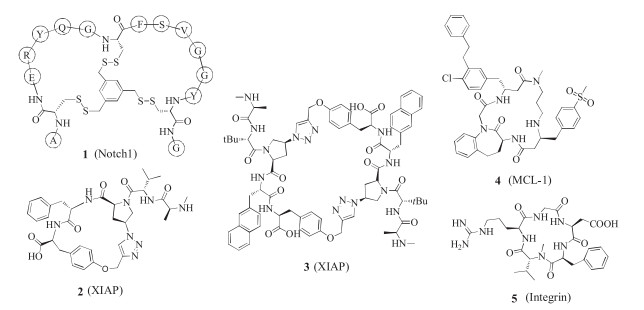
To increase conformational constraint to the displayed peptide, cysteines are often added to both the N- and C-terminus of the displayed sequence, and disulfide bridges are formed before selection. Inspired by natural peptides with multiple disulfide bridges such as cyclotides and conotoxins, researchers designed and synthesized bicyclic phage library by using four cysteines in the peptide library design. Because of the increased constraint on the peptide, bicyclic peptide libraries may facilitate identification of high affinity ligands [26]. However, formation of multiple isomers by different pairing pattern and isomerization by disulfide exchange in systems containing free thiol became an issue in this case. Heinis et al. developed another method to construct bicyclic phage library by using biocompatible thioether formation. The peptide sequences encoded by a phage library contain three invariable cysteines, which react with 1, 3, 5-tris-(bromomethyl) benzene, or other analogs with the same symmetry, to give bicyclic peptides [27, 28]. This method has been applied in discovery of PPI inhibitors for targets such as Notch1 and β-catenin, as well as proteases and others. As an example, bicyclic peptide 1 (Fig. 1) binds to the negative regulation region of Notch1 with the Kd value of 150 nmol/L [29].

|
Download:
|
| Fig. 1. Structures of macrocyclic peptides 1–5. | |
{kind=link}
2.2. Ribosomal display
Cell-free methods like mRNA and ribosomal display enable several orders larger library size than phage display (1014 vs. 109). However with the 20 proteinogenic amino acids, cyclization chemistry is basically limited to cysteines. To increase the structural diversity of the library, non-proteinogenic amino acids need to be incorporated into the expression system with technical feasibility. Suga et al. has reported the use of a ribosomal display method, termed Random non-standard peptides integrated discovery (RaPID), to circumvent this issue [30]. Instead of using aminoacyl t-RNA synthases, they created promiscuous aminoacylating ribozymes, so-called flexizymes, which essentially recognize activated carboxylates, to charge tRNA with amino acids. Flexizymes thus allow extensive genetic code reprogramming with non-canonical amino acids, which can be used for peptide cyclization on the mRNA. This method enables the use of cyclization method that are compatible to the peptide and expression system, for example, 1, 3-dipolar cycloaddition and thioether formation.
2.3. SICLOPPSSplit-intein circular ligation of peptides and proteins (SICLOPPS) is another method for cyclic peptide hit generation, utilizing intein splicing, a process that protein domains called inteins self-excise and link the flanking sequences (exteins) by a native peptide bond. Different from the display technologies that usually use target-based affinity screening, SICLOPPS uses phenotypic screening, in which plasmid library encoding different lengths and sequences of inteins is constructed and transformed into the cells containing a functional assay. After expression within the cell, the inteins form a head-to-tail backbone-linked cyclic peptide with a Cys or Ser in the sequence. The identity of the active cyclic peptides is revealed by isolating the plasmids from cells that show the desired phenotype. SICLOPPS technology has been used in identification of PPI inhibitors of HIF-1α/HIF-1β, AICAR transformylase homodimerization among others, and is reviewed recently [31].
2.4. DNA-encoded chemical libraryDrug screening using DNA-encoded chemical library (DEL) is a technology that combines the ideas of combinatorial chemistry and DNA sequencing. In brief, large combinatorial chemical compound libraries are synthesized in split-and-pool cycles, with each step of structure modification recorded by a DNA sequence, which has the only sequence corresponding to the building block and is covalently linked to the compound (the step named "encoding"). At the end, every compound in the library has its own unique DNA tag. Affinity screening is carried out by applying the library to a protein target of interest, and the DNA tags of the compounds bound to the target are isolated, amplified by PCR, and analyzed by high-throughput DNA-sequencing. Analysis of the sequence recovers the accurate structure information of the hits, and the compounds without DNA-tag are synthesized to confirm the activity. This technology provides much lower detection limit compared to direct structure analysis on the compounds using chemical methods, and provides more chemical diversity compared to biological display technologies [32-34].
Using this technology, Seigal et al. identified macrocyclic peptide antagonists against PPI target XIAP (X-chromosomelinked inhibitor of apoptosis protein) [35]. XIAP plays a key role in apoptosis through binding to pro-apoptotic caspases and prevents programmed cell death. The researchers designed their library based on a known XIAP-binding inhibitor, a tetrapeptide Ala-ValPro-Leu (AVPI) from the N-terminal region of the Smac protein. They used Huisgen 1, 3-dipolar cycloaddition to prepare a P2-P5 cyclized peptide library and screened against both of XIAP's caspase binding domains, BIR2 and BIR3. Moderate affinity ligands of BIR3 were identified, and were confirmed by the synthesis and biophysical study of the macrocyclic peptides without DNA-tag. Cyclic peptide 2 binds to XIAP BIR2 and BIR3 with submicromolar affinity and has an EC50 value of 1.40 μmol/L in in vitro caspase-3 rescue assay. Subsequent screening with P3-P5 library and further optimization afforded homodimers of the peptide with superior XIAP and cIAP affinity and high potency in caspase-3 rescue assay in cell lysate. One of the homodimers 3 also showed high potency in antiproliferation assays with different tumor cell lines and efficacy in human tumor xenograft models in mice, which was not shown by other macrocyclic peptides in the research probably due to poor cell permeability.
Johannes et al. reported the discovery of macrocyclic peptide MCL-1 inhibitors by combination of DEL-screening and medicinal chemistry optimization [36]. MCL-1 is an anti-apoptotic protein from the BCL-2 family. The hit identified by DEL-screening has the scaffold of a tetrapeptide but with non-proteinogenic amino acids for all positions. Optimization was based on the structural information from co-crystalization of this compound with MCL- 1 protein, including macrocyclization between the N- and Cterminus of this tetrapeptide, and further modifications of two side chains. This afforded the final compound 4 with low nanomolar potency in PPI assay and low-micromolar activity in cell-based assay.
3. Structure-based design of macrocyclic peptidesAccording to structure features, the interface of PPIs can be divided into three types [1, 4, 5, 11]. The first is PPI with a primary peptide epitope, i.e., one partner uses 1–4 consecutive amino acids on a chain to interact with the other. Examples of this category include the integrin-binding tripeptide motif Arg-Gly-Asp (RGD) from fibrinogen and others, and the tetrapeptide Ala-Val-Pro-Ile (AVPI) for inhibitor of apoptosis proteins (IAPs) including cIAP1, cIAP2 and XIAP. The second type contains a single unit of secondary structure, such as α-helix, binding to a hydrophobic groove. The hot residues are distributed along the peptide chain, but not consecutively. This type of PPIs is good targets for peptide inhibitors and is under intensive studies. It has been estimated that up to 40% of PPIs belong to this category, in which 60% use an α-helix. Examples include the MDM2/MDMX and p53, BCL family proteins and their partners like BAD and BAK. The third type is PPIs using tertiary structural epitopes, i.e., discontinuous binding sites on several parts of the protein. This type of interface is large and shallow, therefore is difficult for drug discovery. Interleukin-17 and Ras are examples belonging to this category.
Structure-based design of cyclic peptides starts with the native peptide sequence from the PPI interface. When the crystallographic structure of the PPI complex or a ligand-protein complex is known, a special type of peptidomimetic can be selected. There are plenty of peptidomimetics mimicking α-helix, β-turns and β-strands. For the type of PPIs using several consecutive residues as binding epitope, a short constrained cyclic peptide would be suitable. With properly positioned flanking residues and modifications, the orientation of sidechains and conformation of peptide backbone can be finely tuned to fit target binding, as exampled by cilengitide 5 (Fig. 1).
The cyclic peptide cilengitide developed in Kessler's group is a head-to-tail cyclized pentapeptide based on the RGD motif [37, 38], which is the hot spot sequence of αvβ3, αvβ5 and α5β1 integrins. By systematic study of L- to D-amino acid substitution on each of the five residues in the sequence RGDFV, a process named "spatial screening", they identified the peptide with a D-Phe, c(RGDfV), showed increased inhibition in a cell-adhesion assay. Further optimization with N-methyl scan resulted in the more active and selective cilengitide c(RGDf-N(Me)V), which has entered phase Ⅲ clinical trials.
4. Stapled peptide inhibitors of p53-MDM2/MDMX and BCL-2 family protein interactionsFor PPI interface involving an α-helix binding epitope, many approaches to stabilize the α-helix have been developed, with stapling being the most studied. First designed by Verdine and coworkers, peptide stapling uses ring-closing metastasis to form hydrocarbon linker between i, i+4 or i, i+7 residues of an α-helix [39]. This may result in increased affinity to the target, improved cell permeability and increased stability. Typical examples of this group are p53-MDM2/MDMX and interactions among BCL family proteins. Due to their key roles in apoptosis, these are the two most intensively studied groups.
MDM2 and MDMX are two major negative regulators of the tumor suppressor p53 protein, and are overexpressed in many tumors. MDM2 is an E3 ubiquitin protein ligase, which binds to a short α-helix in p53 containing three key hydrophobic residues (F19, W23, and L26) and directs p53 to proteasomal degradation. MDMX, a structural homologue of MDM2 lacking ubiquitin ligase activity, can also sequester p53 for degradation via heterodimerization with MDM2. There are both small molecule and peptide inhibitors in clinical trials, and new structures are being discovered [40-43]. Stapled peptide ALRN-6924 is the most advanced peptidic drug candidate in this group, which is being tested in phase Ⅱ clinical trial. It is a dual inhibitor of p53-MDM2 and the p53-MDMX interactions, and was developed from ATSP-7041 (6, Fig. 2), which was in turn obtained by i, i+7 stapling of the peptide pDI discovered by phage display. SAH-p53-8 (7) is also an i, i+7 stapled peptide using sequence taken from the binding epitope of p53. Both SAHp53-8 (Kd MDM2/MDMX = 55/2.3 nmol/L) and ATSP-7041 (Kd MDM2/MDMX = 0.9/6.8 nmol/L) are dual inhibitors, with ATSP- 7041 being much more potent to MDM2 [44, 45]. In comparison, small molecule p53-MDM2 inhibitor nutlins discovered from highthroughput screening are specific for MDM2, and are also in clinical trials. It will be interesting to see the outcomes of these two different groups of drug candidates.
BCL-2 family proteins include pro-apoptotic members (e.g., BID, BIM, PUMA) that activate BAK and BAX, which are also proapoptotic family members, and anti-apoptotic ones (e.g., BCL-2, BCL-XL, MCL-1) that inhibit these proteins through PPIs. Two other members, BAD and NOXA, are also pro-apoptotic by sequestering the anti-apoptoic members. This PPI network provides abundant potential drug targets for cancer therapy. Both small molecules and peptides are developed to target these proteins, such as palmytoylated dodecapeptide targeting BCL-2 [46]. Although venetoclax (ABT-199), a selective inhibitor of BCL-2, was approved by FDA in 2016, drugs targeting other BCL-2 family members are still lacking. A group of stapled peptides called "stabilized-α-helix of BCL-2 domains" (SAHBs) are developed in Verdine's and Walensky's groups, targeting BID or MCL-1, respectively (MCL-1 SAHBD 8 shown in Fig. 2 as an example). Small molecule and peptide inhibitors to BCL-2 family proteins are summarized in a recent review [47].

|
Download:
|
| Fig. 2. Structures of macrocyclic peptides 6–10. | |
{kind=link}
5. Recent examples of macrocyclic peptide PPI regulators 5.1. Rab GTPases
Rab GTPases are the largest subfamily of small GTPases. Located at the inner side of plasma membrane, they are master regulators of membrane trafficking. As Rab interacts with many effector proteins and have implications in diseases like cancer and neurodegenerative diseases, Rab inhibitors may have potential in both pharmaceutical and basic research purpose. Grossmann, Waldmann and co-workers have reported the discovery of peptidic inhibitors to these difficult targets [48, 49]. The starting sequence was discovered from a search in the Protein Data Bank, looking for α-helices in the PPI interface of Rab family members and their binding partners. A stapled peptide StRIP3 (9), with modified sequence from Rab6-interacting protein 1 (R6IP, aa 900–916), showed low micromolar affinity to the activated (GTP-bound) form of Rab8a (Kd = 30 μmol/L). This stapled peptide showed rapid proteolytic degradation and poor cellular uptake. These researchers then introduced α-methylated amino acids and a second hydrophobic cross-link in combination with a careful sequence optimization, in which the binding affinity retained and the other pharmacological properties were dramatically improved. The resulted double-stapled peptide StRIP16 (10) has increased affinity to the target (Kd = 12.7 μmol/L), and huge improvement in stability compared to the parent peptide StRIP3 (t1/2>3000 vs. 0.3 min). Despite its overall negative net charge, the cellular uptake of StRIP16 is in the range of the cell penetrating peptide Tat49-57. This work provides an elegant example for the improvement of bioactivity and bioavailability of conformationally constrained peptides.
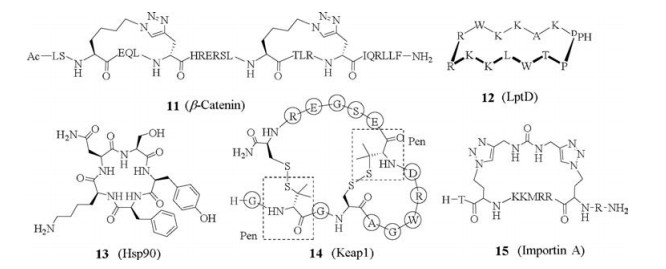
5.2. β-Catenin-BCL9Wang and co-workers reported triazole-stapled peptides targeting interaction between β-catenin and B-cell CLL/lymphoma 9 (BCL9), which is critical for the transcriptional activity of β-catenin [50]. The starting point is a helical segment from BCL9 at the interface, binding to a large groove in β-catenin. The combination of L-Nle(ε-N3) and D-Pra substituted at the i and i+4 positions, respectively, produced the best results for generation of single stapled peptides. Furthermore, double-stapling, which is accessible due to the unsymmetrical nature of Huisgen 1, 3-dipolar cycloaddition, resulted in peptide 11 (Fig. 3) with over 95% helical propencity, improved potency binding to β-catenin, and increased proteolytic stability in cell media.

|
Download:
|
| Fig. 3. Structures of macrocyclic peptides 11–15. | |
{kind=link}
5.3. LptD
LPS-assembly protein LptD (also called Imp or OstA) is an outermembrane protein in Gram-negative bacteria, and functions in the assembly of lipopolysaccharides (LPS) in the outer-membrane. In contrast to most other studies that use well-defined PPI target for affinity screening or rational drug design, in this work the macrocyclic peptide ligands were identified first. The work started with a porcine antimicrobial peptide protegrin Ⅰ (PG-Ⅰ), which adopts a β-hairpin secondary structure in solution. Replacement of the two disulfide bridges in PG-Ⅰ by a D-Pro-L-Pro motif, a known β-turn mimetic, resulted in macrocyclic peptides with improved antimicrobial activity and reduced toxicity. Iterative optimization led to the identification of L27-11 (12) that has nanomolar antimicrobial activity and high specificity against Pseudomonas species including the pathogenic Pseudomonas aeruginosa. Another advantage of L27-11 is that it is not amphiphilic thus without membrane-lytic toxicity. Further optimization resulted in the compound POL7001, in which all arginines of L27-11 were substituted by L-2, 4-diaminobutyric acid, and then a clinical candidate POL7080. Mechanism study discovered the target for these macrocyclic peptides is the outer membrane protein LptD. Binding of the peptides to LptD disrupts complex formation of LptD with another outer membrane-located lipoprotein LptE. [15, 16, 51]
5.4. Hsp90McAlpine and colleagues reported the discovery of cyclic peptide ligands to the C-terminus of heat shock protein 90 (Hsp90) [52-55]. Hsp90 is a chaperone protein involved in the maintenance of protein homeostasis. Hsp90 interacts with over 400 proteins, assisting the folding, intracellular transport, maintenance and degradation. The interactions between Hsp90 and its effector proteins are implicated in human diseases such as cancer. Hsp90 exists as a homodimer, where each monomer contains three domains: (1) The N-terminus containing an ATP-binding site, (2) the middle domain that contains binding sites for client proteins, and (3) the C-terminus that contains binding sites for cochaperones and serves as the dimerization domain. Classical Hsp90 inhibitors target the ATP-binding site located in the Nterminus and activate the heat shock response (HSR), which is responsible for the disappointing results observed with these inhibitors in clinical trials. Inhibiting co-chaperone binding at Hsp90's C-terminus actively decreases the cell protection mechanisms. Based on a 12-amino acid sequence reported by Kawakami and co-workers, McAlpine's group synthesized truncated peptides of 5–8 amino acids long, either from the C-terminus or N-terminus of the 12-mer peptide, and their head-to-tail cyclized analogues. From these peptides, the cyclic pentapeptide 13 from the Cterminal of the 12-mer was found to be the most active, with the IC50 of 4 μmol/L for the inhibition of Hsp90β-Cyp40 interaction, which is more than 10-fold more effective than the original 12-mer linear peptide. Cyclic heptamer and cyclic octamer from the Cterminus were also active, with IC50 values of 15 μmol/L and 18 μmol/L, respectively. All three cyclic peptides were more potent than the orginal 12-mer linear peptides. All the linear truncated peptides were inactive. Further substitution of each residue in this cyclic pentapeptide with an alanine, or lysine, or D-isomer showed that electrostatic interactions were critical for binding between Hsp90 and the effector proteins. Inversion of the original Ala from L- to D-isomer resulted in complete loss of activity. This indicates that an explicit organization of the backbone is critical. And this work also showed that macrocyclic peptides can be used to target parts of the functions of multi-functional proteins.
5.5. Keap1-Nrf2The transcription factor nuclear factor erythroid-derived related factor 2 (Nrf2) plays a key role in antioxidant response of the cell. Kelch-like ECH-associated protein 1 (Keap1) binds to Nrf2 through PPI, keeps it at low level by targeting it to ubiquitinmediated degradation. As cellular oxidative stress is associated with many diseases such as cardiovascular disease, cancer, inflammation and neurodegenerative diseases, this PPI becomes a therapeutically relevant target. Compared to prior work using nonspecific small molecule Michael acceptors to modify cysteines on Keap1, inhibition of Nrf2 degradation by blocking the Nrf2– Keap1 PPI has the potential to be a more selective method of Nrf2 activation. A β-hairpin loop was previously identified as critical for this PPI. A 16-residue peptide from Nrf2 (A69–L85) containing this loop binds Keap1 with a Kd of 24 nmol/L, retaining much of the affinity of full-length Nrf2 (Kd of 5–9 nM) [56].
Lu et al. reported a shorter cyclic peptide that disrupts the PPI of Keap1-Nrf2 [57]. The structure information of Keap1-Nrf2 indicated that a six-residue motif DEETGE on Nrf2 is pivotal for Keap1 binding, and hydrophobic residues adjacent provide additional affinity. The 9-mer containing this motif (Ac-LDEETGEFL-OH) can retain the major part of the binding affinity. For this peptide, the co-crystalized structure showed a 7.4 Å distance between the N- and C-terminus. Therefore the authors added two additional residues, Gly-Gln, to the N-terminus, and prepared the head-to-tail cyclic peptide (c[GQLDPETGEFL]). This peptide showed a significantly improved affinity to Keap1, with a Kd of 18.12 nmol/L, compared to the Kd of 86.96 nmol/L for linear peptide.
Wu and colleagues reported a phage display-based strategy for generation of bicyclic peptide ligands constrained by disulfide bridges, and applied it for ligand screening against Keap1. A phageencoded peptide library with the format CXC(X)5C(X)5C was designed. The CXC motif can direct the oxidative folding of the four-cysteine peptides towards either the bridged bicyclic or the fused bicyclic structure, therefore reduces the number of isomers. By utilizing an orthogonal disulfide formation between penicillamine (Pen) and cysteine [58], where the heterodimer is preferred than the homodimers, bi- or even multi-cyclic peptide structures can be obtained with high selectivity. Using this strategy, they identified bicyclic peptide 14 containing two Cys-Pen pairs binding to Keap1 with the Ki value of 0.28 μmol/L [59].
5.6. HNF1β-Importin αThe transcription factor hepatocyte nuclear factor 1β (HNF1β) is ubiquitously overexpressed in clear cell carcinoma and is seen as an attractive therapeutic target for ovarian, pancreas and certain types of breast cancer. Spring and co-workers reported the development of non-helical constrained peptides targeting the HNF1β-importin α interaction [60]. The nuclear localization sequence (NLS) of HNF1β, TNKKMRRNRFK, was the main interaction motif with importin α. X-ray crystallographic data and molecular dynamics simulations showed that this segment took a non-helical structure. Guided by the structural information, they designed and synthesized macrocyclic peptides with dialkynyl linkers through two-component double-click chemistry. All the constrained peptides were more cell-permeable than the linear HNF1β NLS control peptide, and the cyclic peptide 15 showed higher binding affinity (Kd = 4.54 μmol/L) to importin α.
6. Cell permeability and biological stability of macrocyclic peptidesUnlike small molecule drugs that usually can enter cell by passive diffusion, peptides are originally thought as hydrophilic molecules that could not cross the plasma membrane. However, some exceptions from the nature provided clues for researchers on how to turn a peptide cell permeable, and these are developed into the two major approaches for cellular delivery of peptides.
The natural product cyclosporine A (16, Fig. 4), orally bioavailable and cell permeable, is used as immunosuppressant and anti-inflammation drug. A cyclized undecapeptide with mostly hydrophobic residues and with seven N-methylated amide bonds, cyclosporine A crosses the plasma membrane by passive diffusion. This inspired researchers to develop similar strategy using N-methylation and adding hydrophobic residues to make cyclic peptides cell permeable. However, there is not a simple correlation between lipophilicity and cell-permeability of macrocyclic peptides, and too high lipophilicity may compromise solubility. The conformation of the peptide backbone and the organization of intramolecular hydrogen bonds (IMHBs) play key roles in the membrane permeability. N-Methylation of the amide bonds exposed to solvent in non-polar environment, but not the ones involved in IMHB formation, would improve permeability. This approach is mostly studied with macrocyclic peptides mainly made up of hydrophobic residues. To increase membrane permeability and to keep high target affinity simultaneously will be the key issue in this optimization process [61-66].

|
Download:
|
| Fig. 4. Structures of macrocyclic peptides 16–20. | |
{kind=link}
The other approach uses a cell-penetrating peptide (CPP), usually covalently bonded to the peptide to be delivered, to help the peptide enter the cell. One of the most used CPP is the Tatpeptide, a sequence from the protein trans-activating transcriptional activator of human immunodeficiency virus 1. Although the detailed mechanism of membrane penetration is not completely clarified yet, CPP has already become a well-established technology with numerous in vitro and in vivo applications, as well as a few drug candidates based on this technology in clinical trials [67, 68].
It is now generally accepted that most CPPs enter cells mainly through endocytotic pathways. The efficacy of uptake is sequence dependent, and for the same CPP it is often concentration dependent. The delivery efficiency of cargo molecule to their cytosolic target is then determined by the uptake and the escape from endocytotic compartment. Lack of cell and tissue selectivity is a major drawback of CPPs. The properties of cargo molecules like size and charge, the conjugation site on both cargo and CPP, will affect the outcome of the delivery, and at present this has to be determined by experiments.
Cyclization and other structural modifications including nonnatural amino acid substitution usually increase the stability of peptides against proteases. But exceptions exist also. As an example, In the Rab8a work by Spiegel and Cromm et al., the singly stapled inhibitory peptide showed poor stability in plasma. A second stapling added to the sequence increased the stability tremendously [48, 49].
Other than simply conjugation of a CPP tag to the macrocyclic peptide, Pei and co-workers developed interesting alternative strategies for designing cell permeable macrocyclic peptides based on a cyclic CPP, cyclo(FΦRRRRQ) (Φ = L-2-naphthylalanine), discovered in their group [69]. As an example, a PDZ domain inhibitor that rescues activity of a chloride ion channel CFTR was discovered [70]. PDZ domain requires the binding peptide in extended conformation, and therefore cyclization may reduce or abolish the affinity. The CPP sequence CRRRRF was added to the Nterminus of the ligand sequence WQVTRV, with a Val→Cys mutation in the ligand part, and this cysteine was used to form disulfide bridge with the N-terminal cysteine. The cyclic peptide 17 with disulfide bridge showed no affinity to PDZ domain, but the affinity did recover when the disulfide bond was cleaved by reductive agent. 17 showed cellular uptake, good serum stability, and biological activity in a cell-based assay. As a comparison, cyclic peptide 18 with amidated C-terminus showed no affinity or activity in these assays. This supports the original hypothesis of an unfavorable cyclization, and also provides an example of the application of reversible cyclization by disulfide bridge.
In another work targeting an oncoprotein K-Ras G12V, this group used a bicyclic strategy. They synthesized a combinatorial library of 5.7×106 bicyclic peptides, with degenerate sequence in one ring for target binding, and an invariant CPP in the other ring. The most potent bicyclic peptide 19 identified in this work induces cancer cell apoptosis at low micromolar concentration [71].
Another bicyclic CPP targets the interaction between NEMO and the inhibitor of kB-kinase (IKK). Transcription factor NF-κB controls the expression of many gene products that are involved in inflammatory response, cell proliferation and apoptosis. Aberrant regulation of NF-κB pathway plays a role in cancer, autoimmune diseases, neurodegenerative diseases and others. NFkB essential modifier (NEMO) is a major upstream regulator to this pathway. NEMO binds to and activates IKK, which phosphorylates its substrate, inhibitor of κB (IκB). This leads to the proteasomal degradation of IkB and release of active NF-κB. Pei and co-workers used bicyclic peptides that are made up of two fused rings through reversible disulfide chemistry. One ring is a cyclic CPP, and the other a NEMO-binding sequence [72]. On arriving the reductive cytosol, the two disulfide bridges will be reduced and the linear NEMO-binding sequence will form. The inhibitor 20 has an IC50 value of 3.5 μmol/L in the disruption of NEMO-IKK complex, determined by FRET assay. And the IC50 value is 20 μmol/L in a cellbased TNFα-induced NF-κB activation assay. As a comparison, a linear CPP fused NEMO-binding peptide has IC50 values of 50 μmol/L and 140 μmol/L, respectively in these two assays.
7. ConclusionA fast expansion of peptide inhibitors of PPI targets with therapeutic significance was seen in recent years, especially with intracellular PPIs that were seen as "undruggable" targets previously. Macrocyclic peptide is one of the most promising subgroup due to the possible improvement in target binding, cell permeability and protease stability with these chemical modifications of peptides. Screening methods employing modern biotechnology and structure-based drug design methods developed in recent years are providing powerful tools for peptide drug discovery and development. In this process, iterative medicinal chemistry optimization is still an important route for lead optimization of cyclic peptide drugs. During this process, accurate structure information of ligand-protein complex provides invaluable assistance for further optimization. With the continuous development of versatile bioorthogonal reactions compatible with peptide synthesis, peptide chemists are provided with more versatile tools for cyclization and modification of peptides, thus to obtain more novel structures with desired properties. Therefore we will see more macrocyclic peptide modulators against PPI targets in both basic research and clinical applications, from which drugs for novel therapies may emerge.
AcknowledgmentsThis research was supported by Principle Training Program of Education Department of Sichuan Province (No. 18CZ0042), Fundamental Research Fund of Chengdu University (No. 2081916027), and Undergraduate Innovation Programm of Chengdu University (Nos. CDU_CX_2018250, CDU_CX_2018251).
| [1] |
M.R. Arkin, Y. Tang, Chem. Biol. 21 (2014) 1102-1114. DOI:10.1016/j.chembiol.2014.09.001 |
| [2] |
A.E. Modell, S.L. Blosser, P.S. Arora, Trends Pharmacol. Sci. 37 (2016) 702-713. DOI:10.1016/j.tips.2016.05.008 |
| [3] |
H. Bruzzoni-Giovanelli, V. Alezra, N. Wolff, et al., Drug Discov. Today 23 (2018) 272-285. DOI:10.1016/j.drudis.2017.10.016 |
| [4] |
M. Pelay-Gimeno, A. Glas, O. Koch, T.N. Grossmann, Angew. Chem. Int. Ed. 54 (2015) 8896-8927. DOI:10.1002/anie.201412070 |
| [5] |
L.G. Milroy, T.N. Grossmann, S. Hennig, L. Brunsveld, C. Ottmann, Chem. Rev. 114 (2014) 4695-4748. DOI:10.1021/cr400698c |
| [6] |
A. Mullard, Nat. Rev. Drug Discov. 15 (2016) 147-149. DOI:10.1038/nrd.2016.23 |
| [7] |
N. Tsomaia, Eur. J. Med. Chem. 94 (2015) 459-470. DOI:10.1016/j.ejmech.2015.01.014 |
| [8] |
L. Nevola, E. Giralt, Chem. Commun. 51 (2015) 3302-3315. DOI:10.1039/C4CC08565E |
| [9] |
Z. Wang, X. Ding, C.L. Tian, J.S. Zheng, RSC Adv. 6 (2016) 61599-61609. DOI:10.1039/C6RA13976K |
| [10] |
H. Hou, D.Q. Sun, Prog. Chem. 27 (2015) 1260-1274. |
| [11] |
A.D. Cunningham, N. Qvit, D. Mochly-Rosen, Curr. Opin. Struct. Biol. 44 (2017) 59-66. |
| [12] |
T.A.F. Cardote, A. Ciulli, ChemMedChem 11 (2016) 787-794. DOI:10.1002/cmdc.201500450 |
| [13] |
A. Zorzi, K. Deyle, C. Heinis, Curr. Opin. Chem. Biol. 38 (2017) 24-29. DOI:10.1016/j.cbpa.2017.02.006 |
| [14] |
R.R. Araghi, A.E. Keating, Curr. Opin. Struct. Biol. 39 (2016) 27-38. DOI:10.1016/j.sbi.2016.04.001 |
| [15] |
A. Luther, K. Moehle, E. Chevalier, et al., Curr. Opin. Chem. Biol. 38 (2017) 45-51. DOI:10.1016/j.cbpa.2017.02.004 |
| [16] |
D. Obrecht, E. Chevalier, K. Moehle, et al., Drug Discov. Today:Technol. 9 (2012) 63-69. DOI:10.2174/157016312799304570 |
| [17] |
Y.Y. Huang, Y.L. Jin, R. Zhao, Sci. China Chem. 59 (2016) 1250-1257. DOI:10.1007/s11426-016-0186-x |
| [18] |
D.Y. Wang, X. Qin, H. Zhao, Z.G. Li, Sci. China Chem. 60 (2017) 689-700. DOI:10.1007/s11426-016-9033-y |
| [19] |
Z.M. Wu, S.Z. Liu, X.Z. Cheng, et al., Chin. Chem. Lett. 27 (2016) 1731-1739. DOI:10.1016/j.cclet.2016.04.024 |
| [20] |
H.K. Cui, Y. Guo, Y. He, et al., Angew. Chem. Int. Ed. 52 (2013) 9558-9562. DOI:10.1002/anie.v52.36 |
| [21] |
Y. Guo, D.M. Sun, F.L. Wang, Angew. Chem. Int. Ed. 54 (2015) 14276-14281. DOI:10.1002/anie.201500699 |
| [22] |
F.L. Wang, Y. Guo, S.J. Li, et al., Org. Biomol. Chem. 13 (2015) 6286-6290. DOI:10.1039/C5OB00741K |
| [23] |
M. Schmidt, A. Toplak, P.J.L.M. Quaedflieg, J.H. van Maarseveen, T. Nuijens, Drug Discov. Today:Technol. 26 (2017) 11-16. DOI:10.1016/j.ddtec.2017.11.007 |
| [24] |
Z.M. Wu, S.Z. Liu, X.Z. Cheng, X.R. Zhao, H.F. Hong, Chin. Chem. Lett. 28 (2017) 553-557. DOI:10.1016/j.cclet.2016.11.001 |
| [25] |
M. Goldflam, C.G. Ullman, Front. Chem. 3 (2015) 69. |
| [26] |
S.Y. Chen, I.R. Rebollo, S.A. Buth, et al., J. Am. Chem. Soc. 135 (2013) 6562-6569. DOI:10.1021/ja400461h |
| [27] |
C. Heinis, Nat. Chem. Biol. 10 (2014) 696-698. DOI:10.1038/nchembio.1605 |
| [28] |
K. Deyle, X.D. Kong, C. Heinis, Acc. Chem. Res. 50 (2017) 1866-1874. DOI:10.1021/acs.accounts.7b00184 |
| [29] |
C. Urech-Varenne, F. Radtke, C. Heinis, ChemMedChem 10 (2015) 1754-1761. DOI:10.1002/cmdc.201500261 |
| [30] |
R. Obexer, L.J. Walport, H. Suga, Curr. Opin. Chem. Biol. 38 (2017) 52-61. DOI:10.1016/j.cbpa.2017.02.020 |
| [31] |
A. Tavassoli, Curr. Opin. Chem. Biol. 38 (2017) 30-35. DOI:10.1016/j.cbpa.2017.02.016 |
| [32] |
G. Zimmermann, D. Neri, Drug Discov. Today 21 (2016) 1828-1834. DOI:10.1016/j.drudis.2016.07.013 |
| [33] |
W.H. Connors, S.P. Hale, N.K. Terrett, Curr. Opin. Chem. Biol. 26 (2015) 42-47. DOI:10.1016/j.cbpa.2015.02.004 |
| [34] |
R.A. Goodnow Jr, C.E. Dumelin, A.D. Keefe, Nat. Rev. Drug Disc. 16 (2017) 131-147. DOI:10.1038/nrd.2016.213 |
| [35] |
B.A. Seigal, W.H. Connors, A. Fraley, et al., J. Med. Chem. 58 (2015) 2855-2861. DOI:10.1021/jm501892g |
| [36] |
J.W. Johannes, S. Bates, C. Beigie, et al., ACS Med. Chem. Lett. 8 (2017) 239-244. DOI:10.1021/acsmedchemlett.6b00464 |
| [37] |
C. Mas-Moruno, F. Rechenmacher, H. Kessler, Anti-Can. Agents Med. Chem. 10 (2010) 753-768. DOI:10.2174/187152010794728639 |
| [38] |
J. Chatterjee, F. Rechenmacher, H. Kessler, Angew. Chem. Int. Ed. 52 (2013) 254-269. DOI:10.1002/anie.201205674 |
| [39] |
C.E. Schafmeister, J. Po, G.L. Verdine, J. Am. Chem. Soc. 122 (2000) 5891-5892. DOI:10.1021/ja000563a |
| [40] |
K.K. Hoe, C.S. Verma, D.P. Lane, Nat. Rev. Drug Discov. 13 (2014) 217-236. DOI:10.1038/nrd4236 |
| [41] |
M. Wade, Y.C. Li, G.M. Wahl, Nat. Rev. Cancer 13 (2013) 83-96. DOI:10.1038/nrc3430 |
| [42] |
W.H. Zhou, X.G. Xu, J. Li, et al., Chin. Chem. Lett. 28 (2017) 422-425. DOI:10.1016/j.cclet.2016.09.001 |
| [43] |
A. Burgess, K.M. Chia, S. Haupt, et al., Front. Oncol. 6 (2016) 7. |
| [44] |
Y.S. Chang, B. Gravesb, V. Guerlavais, et al., Proc. Natl. Acad. Sci. U. S. A. 110 (2013) E3445-E3454. DOI:10.1073/pnas.1303002110 |
| [45] |
P.M. Cromm, J. Spiegel, T.N. Grossmann, ACS Chem. Biol. 10 (2015) 1362-1375. DOI:10.1021/cb501020r |
| [46] |
C.L. Zhang, S. Liu, X.C. Liu, J.M. Gao, S.L. Wang, Chin. Chem. Lett. 28 (2017) 1523-1527. DOI:10.1016/j.cclet.2017.03.010 |
| [47] |
A. Ashkenazi, W.J. Fairbrother, J.D. leverson, A.J. Souers, Nat. Rev. Drug. Discov. 16 (2017) 273-284. DOI:10.1038/nrd.2016.253 |
| [48] |
P.M. Cromm, J. Spiegel, P. Kuchler, et al., ACS Chem. Biol. 11 (2016) 2375-2382. DOI:10.1021/acschembio.6b00386 |
| [49] |
J. Spiegel, P.M. Cromm, A. Itzen, et al., Angew. Chem. Int. Ed. 53 (2014) 2498-2503. DOI:10.1002/anie.201308568 |
| [50] |
S.A. Kawamoto, A. Coleska, X. Ran, et al., J. Med. Chem. 55 (2012) 1137-1146. DOI:10.1021/jm201125d |
| [51] |
N. Srinivas, P. Jetter, B.J. Ueberbacher, et al., Science 327 (2010) 1010-1013. DOI:10.1126/science.1182749 |
| [52] |
L.K. Buckton, H. Wahyudi, S.R. McAlpine, Chem. Commun. 52 (2016) 501-504. DOI:10.1039/C5CC03245H |
| [53] |
Y.C. Koay, H. Wahyudi, S.R. McAlpine, Chem. Eur. J. 22 (2016) 18572-18582. DOI:10.1002/chem.v22.51 |
| [54] |
M.N. Rahimi, L.K. Buckton, S.S. Zaiter, et al., ACS Med. Chem. Lett. 9 (2018) 73-77. DOI:10.1021/acsmedchemlett.7b00310 |
| [55] |
Y. Wang, S.R. McAlpine, Chem. Commun. 51 (2015) 1410-1413. DOI:10.1039/C4CC07284G |
| [56] |
J. Gavenonis, B.A. Sheneman, T.R. Siegert, et al., Nat. Chem. Biol. 10 (2014) 714-723. |
| [57] |
M.C. Lu, Q. Jiao, T. Liu, et al., Eur. J. Med. Chem. 143 (2018) 1578-1589. DOI:10.1016/j.ejmech.2017.10.052 |
| [58] |
Y.W. Zheng, L.X. Zhai, Y.B. Zhao, C.L. Wu, J. Am. Chem. Soc. 137 (2015) 15094-15097. DOI:10.1021/jacs.5b10779 |
| [59] |
M.R. Zha, P. Lin, H.W. Yao, Y.B. Zhao, C.L. Wu, Chem. Commun. 54 (2018) 4029-4032. DOI:10.1039/C7CC09142G |
| [60] |
M.M. Wiedmann, Y.S. Tan, Y. Wu, et al., Angew. Chem. Int. Ed. 56 (2017) 524-529. DOI:10.1002/anie.201609427 |
| [61] |
T.R. White, C.M. Renzelman, A.C. Rand, Nat. Chem. Biol. 7 (2011) 810-817. DOI:10.1038/nchembio.664 |
| [62] |
Y.Y. Liang, S. Tang, J.S. Zheng, Prog. Chem. 26 (2014) 1793-1800. |
| [63] |
Y.C. Koay, N.L. Richardson, S.S. Zaiter, ChemMedChem 11 (2016) 881-892. DOI:10.1002/cmdc.201500572 |
| [64] |
B. Over, P. Matsson, C. Tyrchan, et al., Nat. Chem. Biol. 12 (2016) 1065-1074. DOI:10.1038/nchembio.2203 |
| [65] |
P. Matsson, B.C. Doak, B. Over, J. Kihlberg, Adv. Drug Deliv. Rev. 101 (2016) 42-61. DOI:10.1016/j.addr.2016.03.013 |
| [66] |
C.L. Ahlbach, K.W. Lexa, A.T. Bockus, et al., Future Med. Chem. 7 (2015) 2121-2130. DOI:10.4155/fmc.15.78 |
| [67] |
K. Kurrikoff, M. Gestin, Ü. Langel, Exp. Opin. Drug. Deliv. 13 (2016) 373-387. DOI:10.1517/17425247.2016.1125879 |
| [68] |
G. Guidotti, L. Brambilla, D. Rossi, Trends Pharm. Sci. 38 (2017) 406-424. DOI:10.1016/j.tips.2017.01.003 |
| [69] |
Z. Qian, P.G. Dougherty, D.H. Pei, Curr. Opin. Chem. Biol. 38 (2017) 80-86. DOI:10.1016/j.cbpa.2017.03.011 |
| [70] |
Z. Qian, X. Xu, J.F. Amacher, et al., Angew. Chem. Int. Ed. 54 (2015) 5874-5878. DOI:10.1002/anie.201411594 |
| [71] |
T.B. Trinh, P. Upadhyaya, Z. Qian, et al., ACS Comb. Sci. 18 (2016) 75-85. DOI:10.1021/acscombsci.5b00164 |
| [72] |
Z. Qian, C.A. Rhodes, L.C. McCroskey, et al., Angew. Chem. Int. Ed. 56 (2017) 1525-1529. DOI:10.1002/anie.201610888 |