 2018, Vol. 29
2018, Vol. 29 



Antibodies are among the most commonly used and most important tools in the biological sciences for understanding biological processes and studying protein expression, localization and interactions. However, they are also among the most common causes of problems in biological experiments due to the difficulty in sourcing validated and batch-to-batch reproducible antibodies [1, 2]. Among two million antibodies on the market, up to half of them are probably unreliable in recognizing the right targets [2, 3]. Moreover, the use of antibodies also including antibody fragments) inside living cells are limited owing to their poor efficiency of cellular uptake and their instability in reducing environments of the cytoplasms; in addition, antibodies suffer from drawbacks such as limited tissue penetration, high production cost, and potential immunogenicity [4-6]. These problems render an opportunity of developing non-immunoglobulin (non-Ig) scaffold binders (or affinity reagents) with relatively-stable structures and relativelylow molecular weights, mostly between 40 and 100 amino acid residues [7]. These non-Ig affinity reagents include Affibodies, Affimers, DARPins, Affilins, Knottins, and Adnectins and many others [8, 9], which have been useful tools in many antibody-like applications including the immune-like affinity assays for detection of proteins, intracellular regulation of protein function, and localization of target proteins [10, 11]. Despite their usefulness and promising potential in replacing antibodies, non-Ig scaffolds usually consist of a large portion of scaffold residues (primarily α-helices and/or β-sheets in structures) and a small fraction of residues (10–20 residues) complementary to the targets for binding; thus greatly hampering the further miniaturization of non-Ig affinity reagents, which would not only reduce their production costs and immunogenicity, but improve their tissue penetration and cellular membrane permeability.
The demand of further miniaturizing antibody-like affinity reagents leads to rapid development of scaffold-free cyclic and multicyclic peptides, for example, peptides cyclized through amide-bond backbone and small organic crosslinkers [12-17]. These peptides usually possess 5–30 amino acid residues, which lie between non-Ig scaffolds and small molecules in size. Monocyclic peptides are most widely explored to develop minimal affinity reagents for proteins [17-21]. This is largely due to the easiness of designing a single loop mimic and synthesizing peptide monocycles. However, multicyclic peptides (including bicycles, tricycles, etc.) usually exhibit higher binding affinity and specificity to target proteins compared to monocyclic peptides, because they are likely to achieve a higher degree of complementarity in shape with target proteins [22]. In addition, multicyclic peptides are usually more constrained in structure, and thus are usually more resistant to enzymatic hydrolysis compared to monocycles. The challenge of mimicking antibodies using multicyclic peptides lies in the lack of efficient screening strategies and computational design algorithms for de novo selecting or designing high-affinity protein binders that take advantage of two or more loops for the binding. Moreover, novel synthetic strategies that allow the design and facile synthesis of multicyclic peptides with more than two or three loops are still desired.
In this review, we highlight the recent development in mono- and multi-cyclic peptide mimics of antibodies. A brief introduction on the mode of antigen-recognition by antibodies will be first given, as this determines why (multi-)cyclic peptides can be exploited for the design of antibody mimics. We then introduce mono- and multi-cyclic peptide affinity reagents that have recently been developed for use in antibody-like applications. Novel synthetic strategies for multicyclic peptides and promising peptide library screening platforms will also be introduced. Finally, we provide a perspective on the design and screening of multicyclic peptide mimics of antibodies in the future.
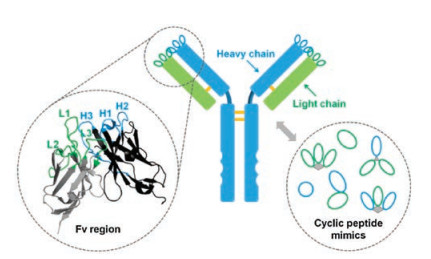
2. Antibody-antigen recognitionA central role of antibodies in humoral immunity is to recognize and bind to a great diversity of foreign invaders (antigens) with high affinity and specificity [23]. In most vertebrates, there are five main classes of antibodies (IgA, IgD, IgE, IgG, and IgM), among which IgGs are the most abundant. IgGs consist of two light chains and two heavy chains (Fig. 1) [24]. Both of them contain variable domains (VL and VH, respectively), which associate together to mediate antigen recognition via six peptide loops known as the complementarity-determining regions (CRDs) (Fig. 1). Other classes of antibodies mainly differ in the heavy chain constant regions (CH); that means the mode of antigen recognition is conserved. Usually, antibodies can specifically bind to their antigens with subnanomolar or lower dissociation constants. How can antibodies recognize the right targets with such a high binding affinity? The answer is given on the "six-loop" structure of their CDRs. The generation or de novo) design of antibodies relies on global optimization of the structures of their six variable loops to achieve binding-competent conformations that precisely complement their targets (Fig. 1). This implies that CRDs of an antibody can be considered as scaffolded multicyclic peptides, rendering the feasibility of designing small peptides with antibody-like affinity and specificity.

|
Download:
|
| Fig. 1. Molecular architecture of an IgG antibody. The Fv region is composed of variable heavy and light domains, each of which has three binding loops mediating antigen recognition (CRDs) (Fv: Protein Data Bank identification number, 3NZ8). Cyclic peptides that mimick the binding of antibodies can take advantage of either a single loop or multiple loops for protein binding. | |
{kind=link}
3. Mono- and multi-cyclic peptide affinity reagents
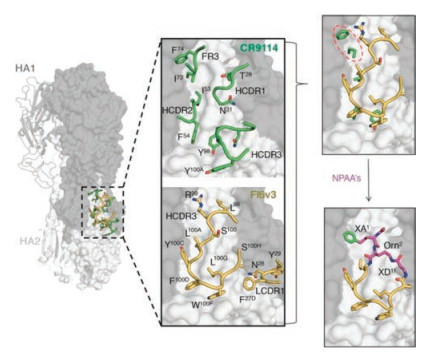
Developing peptide-based affinity reagents with antibody-like affinity and specificity is a long-term dream in the field of peptides and proteins. Cyclic peptides that mimick the binding of antibodies can take advantage of either a single loop or multiple loops for protein recognition. There are decades old reports on cyclic peptide mimics of antibody hypervariable loops, albeit with lower affinity [21]. But the idea planted by those work has indeed inspired generation of novel peptide mimics of antibodies with potent binding affinity. Recently, Kadam et al. reported the design and iterative optimization of a cyclic peptide that targets influenza hemagglutinin (HA) [18]. The iterative workflow used by them for the peptide design involves (Fig. 2): (1) The use of a peptide sequence borrowed from a neutralizing antibody that inhibits HA of influenza A through a single hypervariable loop; (2) Constraining the peptide in the bound conformation through different cyclization chemistries; and (3) Improving the affinity of the peptide binding to HA by extensive introduction of non-natural amino acids. It is worth stressing that the success of this workflow for the design of potent peptide binders greatly benefits from structure information of the antibody-HA complex (Fig. 2). Alternative strategies to create cyclic peptide mimics of antibodies rely on peptide library screening such as phage display and mRNA display [25, 26]. Selection of cyclic peptide binders to proteins by these strategies requires no structural information on complexes of antibodies and antigens (i.e., targets) that currently is largely lacking. Someone might argue that these selected peptide binders may not be strictly recognized as mimics of antibodies, as they were not derived from any specific antibody CRD loop. But we categorize them as mimics of antibodies if these cyclic peptides have antibody-like binding affinity and specificity towards their targets and the binding is mediated by one or more peptide loops. It is no surprise that most monocyclic peptide binders only exhibit relatively weak binding affinity to their targets (many of them with micromolar and a few with nanomolar affinity), but bicyclic peptides with nanomolar or even picomolar affinity have been developed in recent years. Heinis and coworkers have made a great contribution to the development of bicyclic peptide ligands against diverse targets including proteases, receptors, and cytokines [27]. Particularly, bicyclic peptides that can bind some therapeutically interesting targets with picomolar affinity and exceptionally high specificity have been developed, e.g., inhibitors for plasma kallikrein (PK) and coagulation factor XIIa (FXIIa) [28, 29]. It is worth noting that though the screening of bicyclic peptide libraries can usually yield binders with affinities in the nanomolar range, the antibody-like picomolar binding affinity and high binding specificity can only be obtained via further structural modification of the selected peptides.

|
Download:
|
| Fig. 2. Design strategy for cyclic peptide mimics of broadly neutralizing antiboies (bnAbs). Structures of a HA-interacting loop from bnAbs bound to the HA stem epitope allow for the design of small cyclic peptides that bound at the binding epitope. Figure copied with permission [18]. Copyright 2017, The American Association for the Advancement of Science. | |
{kind=link}
4. Synthetic methods for multicyclic peptides
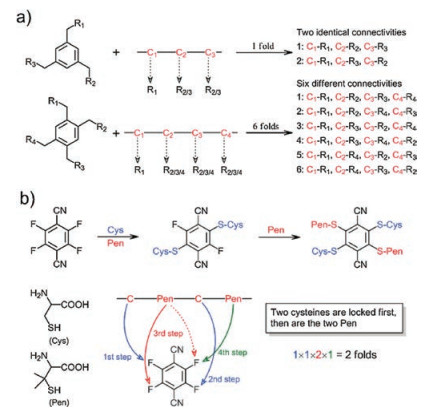
Synthetic methods for multicyclic peptides are the prerequisite for developing antibody-like peptides that bind their targets with two or more loops. For the synthesis of bicyclic peptides, one of the most concise methods should be the cyclization of a peptide with three cysteine residues via an organic crosslinker containing three symmetric thiol-reactive groups [30, 31]. This method is exceptionally elegant, because it can be used for cyclizing unprotected peptides in aqueous solvents at room temperature, and importantly it has been extensively used for the post-translational synthesis of bicyclic peptide libraries. Many other methods for peptide bicyclizations have been developed, including ring-closing metathesis, metal-catalyzed C-C coupling, and azide-alkyne cycloaddition, which usually require the introduction of paired unnatural amino acid residues in peptides [32-35]. In addition, bicyclic peptides can be obtained by sequential cyclizations with two bifunctional crosslinkers [36-38]. In contrast, tricyclic peptides are relatively difficult to be designed and synthesized, though existing synthetic methods for bicyclic peptides can in principle be applied, solely or in combination, to tricyclic peptide synthesis; because this would result in either remarkable increase in complexity of the synthetic procedures or inevitable implication of many regioisomers upon the cyclizations [39, 40]. For example, while an organic crosslinker with four thiol-reactive groups is used to cyclize a peptide with four cysteine residues, a mixture of at least six regioisomers will be formed in theory after the cyclizations (Fig. 3). Novel strategies that can take advantage of the concise thioether bond formation for cyclizations without involving the formation of regioisomers are greatly desired. Recently, Liu et al. have reported the discovery of a small phenyl molecule with four isosteric thiol-reactive groups of sequentially varies reactivity [41]. This molecule can be exploited in combination with cysteine/penicillamine thiolates of different nucleophilic reactivity for precisely regulated and one-pot locking (PROP locking) of linear peptides into specific tricyclic structures (Fig. 3). This PROP-locking strategy relies on multistep and sequential thiolate/fluorine nucleophilic substitutions, which are not only rapid but high specific, thus enabling efficient locking of peptides with high amino acid diversities without protecting groups [41, 42]. However, the PROP-locking of peptides still results in formation of two regioisomers, thus a facile and more advanced strategy that can cyclize peptides into a single tricyclic structure remains to be invented.

|
Download:
|
| Fig. 3. a) Schematic illustration of the reaction of organic scaffolds bearing three and four symmetric thiol-reactive groups with peptides containing three and four cysteine residues; note that, six possible regioisomers can be formed after the tether of the four-cysteine-containing peptide. b) PROP-locking of a peptide with two cysteine and two penicillamine (Pen) residues by 4F-2CN; only two expected regioisomers can be formed. Reproduced with permission [41]. Copyright 2017, Wiley-VCH. | |
{kind=link}
5. Screening platforms
Biosynthetic peptide libraries have played an important role in de novo discovery of peptide affinity reagents, including phage, ribosomal, and mRNA display, among which phage display is playing the most prominent role to date [43, 44]. In phage display, encoded random peptides are displayed onto the phage surface coat proteins [45]. After display, phages bearing the encoded peptides can be selected based on affinity of the displayed peptide for an immobilized protein target (Fig. 4). Phage displayed monocyclic peptide libraries consisting of random peptides constrained with a disulfide bridge, including the Ph.D.-C7C library that is distributed commercially by New England Biolabs [46], have been routinely selected for a range of diverse targets. Interestingly, phage display peptide libraries bearing cysteine residues can also be chemically modified by reactions with thiolate-reactive crosslinkers to generate monocyclic or bicyclic peptide libraries (Fig. 4). For example, a bicyclic peptide library can be created by cyclization of a peptide library of the format ACX6CX6CG with 1, 3, 5-tris (bromomethyl)benzene [13]. To date, a number of bicyclic peptide ligands selected from these phage display libraries are increasingly used as either biological tools or therapeutic leads [27]. mRNA display represents another screening platform of rapid development [47]. In particular, mRNA display allows the incorporation of reactive unnatural amino acids into random peptides for cyclization (Fig. 4) [48]. Recently, mRNA displayed mono- and multicyclic peptide libraries have been extensively used for the selection of peptide binders against a range of diverse targets [26]. As the library size of mRNA display is several orders of magnitude larger than that of phage display, peptide ligands selected from mRNA displayed libraries usually exhibit relatively higher affinity and specificity for targets. However, further optimization of peptide sequences or structures may still be required to achieve antibodylike affinity in the subnanomolar range.

|
Download:
|
| Fig. 4. a) Selection of peptide binders to target proteins through phage display technology; cyclization of displayed peptides on phages. b) Cyclic peptide libraries with incorporated unnatural residues generated from a ribosome-expressed system. | |
{kind=link}
6. Conclusions and outlook
Cyclic peptide mimics of antibodies can take advantage of the high binding affinity and specificity of antibodies, but overcoming some of their drawbacks. For example, cyclic peptides are much smaller in size than antibodies, thus allowing them to penetrate deeper into tissues and seek out binding sites that antibodies cannot reach [49]. In addition, cyclic peptides are much easier and cheaper to produce than antibodies and can usually be directly synthesized using solid phase peptide synthesis (SPPS). To date, almost all of the cyclic peptide affinity reagents were developed based on either monocyclic or bicyclic scaffolds. However, as most antibodies bind their targets with three or more loops to achieve a more precise complementarity with the epitope and thus higher binding affinity and specificity [24], the generation of higher affinity peptide binders might rely on the technological development of grafting multiple (antibody-)loops onto multicyclic peptide scaffolds. This would greatly benefit from recent developments on novel screening platforms and computational design algorithms [48, 50]. For example, new computational methods have enabled the de novo design of structurally diverse disulfidecrosslinked peptides and mini-protein binders of roughly 40 residues to targets such as influenza haemagglutinin and botulinum neurotoxin B [51, 52]. It should be no surprise in the near future that peptide affinity reagents mimicking multiple loops of an antibody will be de novo created computationally. Novel synthetic methods that are amenable to biosynthetic peptide library design will also play a role in accelerating the discovery of novel multicyclic peptide binders, particularly new organic crosslinkers that allows the multiple cyclizations of unprotected peptides under mild conditions in one-pot cascade reactions. Finally, small peptide binders might be quickly filtered out of the body, thus an extension to make them more antibody-like will be of particular interest to in vivo applications. Possible routes include engineering them to bind reversibly albumin (or other serum proteins with long circulating half-lives) or mimicking simultaneously the ability of antibodies to target Fc gamma receptors (FcγRs) [53, 54].
AcknowledgmentsWe would like to acknowledge the financial support from the National Natural Science Foundation of China (No. 21475109), the Program for Changjiang Scholars and Innovative Research Team in University (No. IRT13036), the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (No. 21521004).
| [1] |
A. Bradbury, A. Pluckthun, Nature 518 (2015) 27-29. DOI:10.1038/518027a |
| [2] |
M. Baker, Nature 521 (2015) 274-276. DOI:10.1038/521274a |
| [3] |
J. Bordeaux, A.W. Welsh, S. Agarwal, et al., Biotechniques 48 (2010) 197-209. DOI:10.2144/000113382 |
| [4] |
A.L.J. Marschall, S. Dubel, T. Boldicke, Mabs 7 (2015) 1010-1035. DOI:10.1080/19420862.2015.1076601 |
| [5] |
M. Akishiba, T. Takeuchi, Y. Kawaguchi, et al., Nat. Chem. 9 (2017) 751-761. DOI:10.1038/nchem.2779 |
| [6] |
P. Holliger, P.J. Hudson, Nat. Biotechnol. 23 (2005) 1126-1136. DOI:10.1038/nbt1142 |
| [7] |
H.K. Binz, P. Amstutz, A. Pluckthun, Nat. Biotechnol. 23 (2005) 1257-1268. DOI:10.1038/nbt1127 |
| [8] |
J. Lofblom, F.Y. Frejd, S. Stahl, Curr. Opin. Biotech. 22 (2011) 843-848. DOI:10.1016/j.copbio.2011.06.002 |
| [9] |
T. Wurch, A. Pierre, S. Depil, Trends Biotechnol. 30 (2012) 575-582. DOI:10.1016/j.tibtech.2012.07.006 |
| [10] |
K. Skrlec, B. Strukelj, A. Berlec, Trends Biotechnol. 33 (2015) 408-418. DOI:10.1016/j.tibtech.2015.03.012 |
| [11] |
C. Tiede, R. Bedford, S.J. Heseltine, et al., Elife 6 (2017) e24903. |
| [12] |
C.J. White, A.K. Yudin, Nat. Chem. 3 (2011) 509-524. DOI:10.1038/nchem.1062 |
| [13] |
C. Heinis, T. Rutherford, S. Freund, G. Winter, Nat. Chem. Biol. 5 (2009) 502-507. DOI:10.1038/nchembio.184 |
| [14] |
Y. Hamuro, M.C. Calama, H.S. Park, A.D. Hamilton, Angew. Chem. Int. Ed. 36 (1997) 2680-2683. DOI:10.1002/(ISSN)1521-3773 |
| [15] |
H.K. Cui, Y. Guo, Y. He, et al., Angew. Chem. Int. Ed. 52 (2013) 9558-9562. DOI:10.1002/anie.v52.36 |
| [16] |
K. Hu, H. Geng, Q.Z. Zhang, et al., Angew. Chem. Int. Ed. 55 (2016) 8013-8017. DOI:10.1002/anie.201602806 |
| [17] |
T.A. Hill, N.E. Shepherd, F. Diness, D.P. Fairlie, Angew. Chem. Int. Ed. 53 (2014) 13020-13041. DOI:10.1002/anie.201401058 |
| [18] |
R.U. Kadam, J. Juraszek, B. Brandenburg, et al., Science 358 (2017) 496-502. DOI:10.1126/science.aan0516 |
| [19] |
T.A. Whitehead, Science 358 (2017) 450-451. DOI:10.1126/science.aap9608 |
| [20] |
M. Favre, K. Moehle, L.Y. Jiang, B. Pfeiffer, J.A. Robinson, J. Am. Chem. Soc. 121 (1999) 2679-2685. DOI:10.1021/ja984016p |
| [21] |
W.V. Williams, T. Kieberemmons, J. Vonfeldt, M.I. Greene, D.B. Weiner, J. Biol. Chem. 266 (1991) 5182-5190. |
| [22] |
S.Y. Chen, I.R. Rebollo, S.A. Buth, et al., J. Am. Chem. Soc. 135 (2013) 6562-6569. DOI:10.1021/ja400461h |
| [23] |
M.J. Feige, L.M. Hendershot, J. Buchner, Trends Biochem. Sci. 35 (2010) 189-198. DOI:10.1016/j.tibs.2009.11.005 |
| [24] |
K.E. Tiller, P.M. Tessier, Annu. Rev. Biomed. Eng. 17 (2015) 191-216. DOI:10.1146/annurev-bioeng-071114-040733 |
| [25] |
I.R. Rebollo, C. Heinis, Methods 60 (2013) 46-54. DOI:10.1016/j.ymeth.2012.12.008 |
| [26] |
R. Maini, S. Umemoto, H. Suga, Curr. Opin. Chem. Biol. 34 (2016) 44-52. DOI:10.1016/j.cbpa.2016.06.006 |
| [27] |
K. Deyle, X.D. Kong, C. Heinis, Acc. Chem. Res. 50 (2017) 1866-1874. DOI:10.1021/acs.accounts.7b00184 |
| [28] |
J. Wilbs, S.J. Middendorp, C. Heinis, ChemBioChem 17 (2016) 2299-2303. DOI:10.1002/cbic.v17.24 |
| [29] |
V. Baeriswyl, H. Rapley, L. Pollaro, et al., ChemMedChem 7 (2012) 1173-1176. DOI:10.1002/cmdc.v7.7 |
| [30] |
P. Timmerman, J. Beld, W.C. Puijk, R.H. Meloen, ChemBioChem 6 (2005) 821-824. DOI:10.1002/cbic.v6:5 |
| [31] |
S.Y. Chen, D. Bertoldo, A. Angelini, F. Pojer, C. Heinis, Angew. Chem. Int. Ed. 53 (2014) 1602-1606. DOI:10.1002/anie.201309459 |
| [32] |
C.E. Schafmeister, J. Po, G.L. Verdine, J. Am. Chem. Soc. 122 (2000) 5891-5892. DOI:10.1021/ja000563a |
| [33] |
L. Mendive-Tapia, S. Preciado, J. Garcia, et al., Nat. Commun. 6 (2015) 7160. DOI:10.1038/ncomms8160 |
| [34] |
Y.H. Lau, Y.T. Wu, M. Rossmann, et al., Angew. Chem. Int. Ed. 54 (2015) 15410-15413. DOI:10.1002/anie.201508416 |
| [35] |
G.J. Hilinski, Y.W. Kim, J. Hong, et al., J. Am. Chem. Soc. 136 (2014) 12314-12322. DOI:10.1021/ja505141j |
| [36] |
H. Jo, N. Meinhardt, Y.B. Wu, et al., J. Am. Chem. Soc. 134 (2012) 17704-17713. DOI:10.1021/ja307599z |
| [37] |
Y.X. Wang, D.H.C. Chou, Angew. Chem. Int. Ed. 54 (2015) 10931-10934. DOI:10.1002/anie.201503975 |
| [38] |
C.M.B.K. Kourra, N. Cramer, Chem. Sci. 7 (2016) 7007-7012. DOI:10.1039/C6SC02285E |
| [39] |
K. Chua, E. Fung, E.D. Micewicz, et al., Angew. Chem. Int. Ed. 57 (2018) 501-505. DOI:10.1002/anie.201709127 |
| [40] |
W.D. Liu, Y.W. Zheng, X.D. Kong, et al., Angew. Chem. Int. Ed. 56 (2017) 4458-4463. DOI:10.1002/anie.201610942 |
| [41] |
J.H. Wang, M.R. Zha, Q.R. Fei, et al., Chem. Eur. J. 23 (2017) 15150-15155. DOI:10.1002/chem.201703139 |
| [42] |
A.M. Davis, A.T. Plowright, E. Valeur, Nat. Rev. Drug Discov. 16 (2017) 681-698. DOI:10.1038/nrd.2017.146 |
| [43] |
A.R.M. Bradbury, S. Sidhu, S. Dubel, J. McCafferty, Nat. Biotechnol. 29 (2011) 245-254. DOI:10.1038/nbt.1791 |
| [44] |
C. Heinis, G. Winter, Curr. Opin. Chem. Biol. 26 (2015) 89-98. DOI:10.1016/j.cbpa.2015.02.008 |
| [45] |
K.A. Noren, C.J. Noren, Methods 23 (2001) 169-178. DOI:10.1006/meth.2000.1118 |
| [46] |
A. Galan, L. Comor, A. Horvatic, et al., Mol. Biosyst. 12 (2016) 2342-2358. DOI:10.1039/C6MB00219F |
| [47] |
Y. Yamagishi, I. Shoji, S. Miyagawa, et al., Chem. Biol. 18 (2011) 1562-1570. DOI:10.1016/j.chembiol.2011.09.013 |
| [48] |
B. Owens, Nat. Biotechnol. 35 (2017) 602-603. DOI:10.1038/nbt0717-602 |
| [49] |
M.L. Azoitei, B.E. Correia, Y.E.A. Ban, et al., Science 334 (2011) 373-376. DOI:10.1126/science.1209368 |
| [50] |
G. Bhardwaj, V.K. Mulligan, C.D. Bahl, et al., Nature 538 (2016) 329-335. DOI:10.1038/nature19791 |
| [51] |
A. Chevalier, D.A. Silva, G.J. Rocklin, et al., Nature 550 (2017) 74-79. |
| [52] |
S.C. Penchala, M.R. Miller, A. Pal, et al., Nat. Chem. Biol. 11 (2015) 793-798. DOI:10.1038/nchembio.1907 |
| [53] |
C. Rader, Nature 518 (2015) 38-39. DOI:10.1038/518038a |