 2018, Vol. 29
2018, Vol. 29 



Photo-caged peptides are peptides that contain photo-protecting groups (PPGs) at the side-chain or termini, which can temporarily mask the original bioactivities of the peptides [1, 2]. Irradiation of photo-caged peptides with the light of the appropriate wavelength removes the PPG and regenerates the native functional group in the peptides. Compared with conventional protecting groups such as acid/base- or redox-sensitive protecting groups, light is a traceless agent that allows essentially "reagent-free" deprotection under a mild condition. This feature has provided synthetic chemists a green alternative of protection strategy and illuminated chemical biologists in various applications. Besides the traceless property of the stimulant, the lightinduced release of functional biomolecules can be controlled precisely in a temporal and spatial manner. With these unique properties, photo-caging strategy is widely used in chemical biology on small molecules as well as biomolecules including nucleic acids, neurotransmitters, and proteins [2-6].
This mini-review focuses on the photocaging of peptides. We start with the introduction of common types of PPGs that have been used in peptide photocaging. Then, we systematically review the methodologies in photocaging the N- and C-terminus, the functional groups on the side chains of natural amino acids, and the unique applications of PPGs in the synthesis of cyclic peptides and native chemical ligation. Lastly, we give some examples to display the recent applications of photocaged peptide probes interactions, protein misfolding, post-translational modifications and peptide drug delivery.
2. The chemistry of peptide photocagingSeveral common features are shared among most PPGs [2, 7]. Yet for peptide photocaging, additional restrictions should be imposed. First, the PPG should be chemically stable in aqueous and organic solvents, and it should endure repeated treatment by reagents used for peptide synthesis if the PPG group is to be installed during solid phase peptide synthesis. Second, the light required for photo-uncaging should be longer than 300 nm. Because the amide bonds and some of the amino acid side chains absorb strongly from 190 nm to 280 nm [8], the photo-uncaging process of a peptide using UV light may be affected by the intrinsic absorption of the peptide structure. Third, the byproduct(s) of the photo-uncaging reaction shall be biocompatible with proteins, cells or tissues. Last, a rapid photo-uncaging kinetics is necessary, as cells or tissues may be damaged upon prolonged illumination under strong light. Due to the space limit, the photo-uncaging kinetics is not included here, and great reviews are available for interested readers [7]. As we believe that the basic understanding of the photo-uncaging mechanisms is the sine qua non of a successful peptide photocaging reaction, here we start from a brief review of several PPGs that can be used in peptide photocaging.
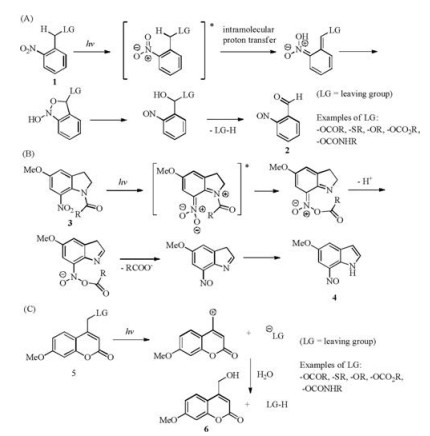
2.1. Commonly used PPGs: 2-nitrobenzyl-type (NB type), 7- nitroindolin-1-yl-type (NI type) and coumarin-4-yl typeThe PPG group containing 2-nitrobenzyl (NB type) 1 has been used in organic synthesis since 1970 [9], and still popularly used due to its clearly illustrated mechanism, the rapid un-caging kinetics and the facile chemistry for its installation. The uncaging mechanism leads to the formation of nitrosobenzaldehyde 2 as the by-product (Scheme 1A) [10]. 7-Nitroindolin-1-yl (NI)-type PPGs 3 are related to NB-type PPGs in structure and share the similar uncaging mechanism with the release of 7-nitrosoindole 4 (Scheme 1B) [11]. The photo-uncaging of coumarin-4-yl-type PPGs 5 is also rapid and can be achieved with light in the visible region with the release of the fluorescent molecule 6 (Scheme 1C) [12]. The NB type of PPGs shall be used with caution for intracellular photolysis, as the byproduct nitrosobenzaldehyde molecule 2 is cytotoxic.

|
Download:
|
| Scheme 1. The photo-uncaging mechanism of NB-type (A), NI-type (B), and Coumarin-4-yl type (C) of PPGs. | |
2.2. The synthesis of photo-caged peptides
Peptidic functional groups, including carboxylic acid, amine, thiol, amide, guanidine, phenol and alcohol, can be photo-caged with PPGs (examples shown in Table 1). Installation of side-chainphotocaged amino acid in the synthetic peptide can normally be achieved through three strategies: (Ⅰ) Incorporation of "Fmoccompatible" photo-caged amino acids during Fmoc solid phase peptide synthesis, (Ⅱ) solution-phase caging of functional groups, and (Ⅲ), on-resin caging of functional groups (Table 1).
|
|
Table 1 Reactions used for photo-caging of functional groups in peptides. |

{kind=link}
2.3. Photo-cleavable cyclic peptides
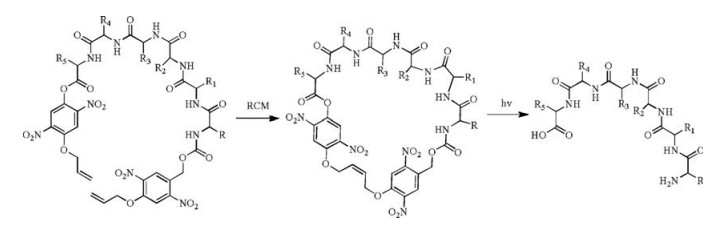
Besides the side-chain protection by PPGs, the termini of the peptide backbone can also be protected [24]. This strategy is illustrated in an example of photo-cleavable cyclic peptides. Peptide cyclization can be achieved by head-to-tail amide formation, chemical ligations, thioalkylation or disulfide formation of cysteine-containing peptides, and ring-closing metathesis (RCM) through allyl groups, and many other methods [25-28]. Peptides in conformationally constrained cyclic form is more stable during circulation and possesses higher membrane permeability to enter the cells, but the cyclic form often compromises the binding affinity towards the intracellular protein target. Hoffmann and Kazmaier proposed a photolytic linearization strategy in which the N- and C-termini of a peptide were linked by an NB-type of the photosensitive linker and cyclized through RCM and photolysis linearizes the peptide (Scheme 2) [13]. This strategy permits photo-responsive conformational control of the peptide.

|
Download:
|
| Scheme 2. A photo-cleavable cyclic peptide | |
{kind=link}
2.4. Photo-caging in peptide ligation
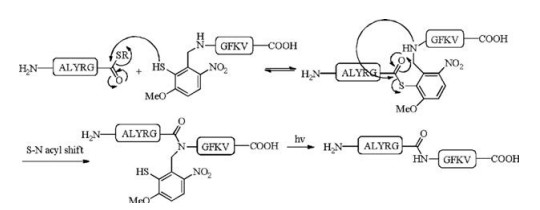
Peptide photocaging has also proven useful in peptide ligation. The traditional native chemical ligation (NCL) requires the presence of an N-terminal cysteine residue and a C-terminal thioester to give an internal cysteine [29-31]. As cysteine is a low abundance amino acid, cysteine-free ligations aim to overcome this restriction. Successful methods include cysteine desulfurization [32-38], serine/threonine ligation (STL) [39-42] and others. Photo-caging provides an alternative: an auxiliary-assisted ligation strategy was developed by using 3-methoxy-6-nitro-2-mercaptobenzaldehyde as the auxiliary to cage the N-terminus of the peptide segment [17, 43]. The auxiliary was designed based on the combination of an NB-type PPG and an acid-labile 2-mercaptobenzyl moiety that can serve the role as an N-terminal cysteine residue to conduct the ligation and acyl-transfer. An example is summarized in Scheme 3 and the ligation reaction proceeded with 91% conversion. After the ligation, irradiation with UV light (365 nm) allowed complete removal of the auxiliary in, without the use of any harsh removal condition.

|
Download:
|
| Scheme 3. Photo-removable auxiliary-assisted chemical ligation. Ligation condition: 6 mol/L GuHCl, 100 mmol/L NaH2PO4, 20 mmol/L TCEP, pH 7.6, R = (4-mercaptophenyl)- acetic acid. | |
{kind=link}
3. Photo-caged peptides in biological studies 3.1. Photo-caged peptides to study protein aggregation
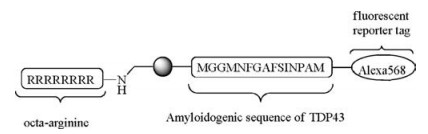
One of the biggest obstacles in the mechanistic studies of protein misfolding and amyloidosis is an ex vivo trigger to induce a rapid protein aggregation with minimal disturbance. A photoresponsible reaction may provide an ideal solution. In a recent example, a photo-caged fluorescent peptide probe mimicking Cterminal of TDP-43, an amyloidogenic peptide was prepared (Scheme 4) [44]. The peptide self-assembled into spherical vesicles in the aqueous medium due to their amphiphilicity. Upon irradiation with UV light, the spherical vesicles dissembled and formed fibrillary structures within seconds. When this strategy was extended to study the aggregation of TDP-43 in mouse neuroblastoma N2A cells, it was observed that uncaged peptide probe recapitulated the seeding of endogenous TDP-43 into aggregates in the cytosol, and cytosolic aggregation of TDP-43 is a pathological hallmark of amyotrophic lateral sclerosis (ALS). This work showcases a novel use of cell-penetrating photo-caged peptide probe in biology.

|
Download:
|
| Scheme 4. A photo-caged fluorescent peptide for photo-triggered nanofibril formation in cells. | |
{kind=link}
3.2. Photo-caged peptides to modulate protein interactions
One of the major applications of photo-caged peptides is to modulate protein-protein interactions. Protecting one of the key residues in the peptide/protein ligands will cause steric hindrance to block such interaction and silence the cellular signal, whereas such a steric block can be removed by photo-decaging to induce the signal transduction [45]. For example, Janett et al. synthetized a photocaged FMRFa peptide, based on the sequence of FMRFa (an agonist peptide that binds to the neurotransmitter receptor MrgR and mimics glutamate-induced ACE in astrocytes) [19]. Photo-caged FMRFa did not lead to elevation of intracellular calcium level in astrocytes. ACE was elicited only when the photo-caged FMRFa peptides were irradiated with UV flashes (365 nm). This shows a successful application of photocaged peptides as neurotransmitters for specific targeting of astrocytes and spatiotemporal control of PAP.
In another example, Chen et al. synthesized two-photonactivatable chemokine CCL5 using photocaged serine through efficient one-pot total chemical synthesis. The chemical groups at serine residues successfully caged the hCCL5 activity, which allowed a spatiotemporally controlled photoactivation of the chemokine activity [22]. More recently, the same group synthesized the first photo-activatable protein antigen to study antigenantibody interaction mediated responses in B cells by screening a set of photo-protected residues such as lysine [23].
3.3. Photocaged peptides as for light-triggered deliveryCaging the cationic groups in cell-penetrating peptides will allow photo-triggered delivery of cargoes into a cell, based on the principle that the cationic charges are responsible for the majority of the cell-penetrating capability [46-48]. For example, Shamay et al. designed a hybrid material with FITC and a proapoptotic peptide, KLAK, conjugated to two CPPs derived from Penetratin separately, through N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer scaffold (Scheme 5) [16]. The positively charged lysine residues were protected with NB-type PPG to mask the cellpenetrating property. After irradiation with UV light for 10 min and a further incubation of 2 h, the fluorescence signal inside cells increased 10-fold compared to that of the caged CPP without irradiation, and a subsequent apoptotic effect cell death of a human prostate cancer cell line) was observed, indicating successful delivery of KLAC-conjugated polymer into the cells. This inspiring experiment points to the direction that light may serve as a physical trigger for controlled release through the change of the cell-penetrating peptide moiety.

|
Download:
|
| Scheme 5. A hybrid material containing proapoptotic peptides and photo-caged cell penetrating peptide on HPMA polymer backbone for photo-controlled cell entry. | |
{kind=link}
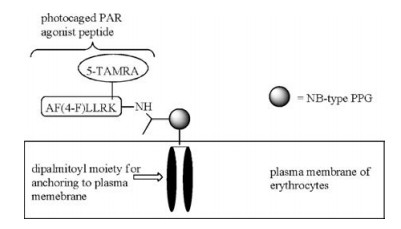
One limitation of peptide-based therapeutics is that peptides undergo rapid proteolysis in blood. To overcome this limitation, O'Banion, Nguyen, and others proposed the use of plasma membrane of human red blood cells as a carrier (or "reservoir") of peptide drugs (Scheme 6) [14, 49]. Through a PPG-derived linker, the peptide drug is conjugated to two lipid chains to allow the anchoring of the lipid-peptide conjugate to the plasma membrane of red blood cells. Utilizing lipid pools as photolabile "protecting group" carries several advantages: first, the surface of red blood cells is covered with glycocalyx, protecting the peptide drugs on the cell surface from proteases. The peptide drugs being carried on the membrane without the need of entering the cytoplasm of "carriers" (red blood cells) and the need of being released. The long circulatory time of red blood cells ensures a slow and controllable release of the drug, and allows the drug to reach all parts of the body through circulation. Together with the precise spatiotemporal control by light irradiation, this presents a universal strategy for the controlled release of peptide therapeutics. For example, a photo-caged agonist peptide of protease-activated receptor (PAR) with a di-palmitoylated moiety was anchored to the plasma membrane of red blood cells. The peptide remained intact after 2 of incubation with proteases, confirming the effect of "protective shielding" by the surface of red blood cells [49]. Photolytic release of functional peptides from the carriers presents a potent method for clinical use.

|
Download:
|
| Scheme 6. A photo-caged PAR agonist peptide using plasma membrane of erythrocytes as a "reservoir". | |
{kind=link}
4. Conclusion
In this review, we aim to bring to the readers a brief account of the chemistry of peptide photocaging and recent examples of photocaged peptides in chemical biology and biomedical science. Notwithstanding that much is known about photochemical reactions, peptide photocaging is still largely under-explored, and it therefore remains an exciting frontier in peptide chemistry. The advancement of optical technology such as two-photon technology will bring new momentum to the development of this field, allowing uncaging in the "phototherapeutic window" (650–1000 nm) [50, 51]. The development of peptide chemistry, optical technology, and protein and cell biology will bring a bright future for the optical regulation of the chemical and biological process, and ultimately leads to new understanding of biological processes and even clinical applications.
AcknowledgmentFinancial support for this work came from the University Grants Committee of Hong Kong (GRF Nos. 14321116 and 14306317, and AoE/M-09/12).
| [1] |
Q. Shao, B. Xing, Chem. Soc. Rev. 39 (2010) 2835-2846. DOI:10.1039/b915574k |
| [2] |
N. Ankenbruk, T. Courtney, Y. Naro, A. Deiters, Angew. Chem. Int. Ed. 57 (2018) 2768-2798. DOI:10.1002/anie.201700171 |
| [3] |
C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer, A. Heckel, Angew. Chem. Int. Ed. 51 (2012) 8446-8476. DOI:10.1002/anie.201202134 |
| [4] |
G. Mayer, A. Heckel, Angew. Chem. Int. Ed. 45 (2006) 4900-4921. DOI:10.1002/(ISSN)1521-3773 |
| [5] |
S. Kantevari, M. Matsuzaki, Y. Kanemoto, H. Kasai, G.C.R. Ellis-Davies, Nat. Methods 7 (2010) 123-125. DOI:10.1038/nmeth.1413 |
| [6] |
D.K. Sinha, P. Neveu, N. Gagey, et al., ChemBioChem 11 (2010) 653-663. DOI:10.1002/cbic.201000008 |
| [7] |
P. Kla'n, T. Solomekm, C.G. Bochet, A. Blanc, R. Givens, M. Rubina, et al., Chem. Rev. 113 (2013) 119-191. DOI:10.1021/cr300177k |
| [8] |
A.R. Goldfarb, L.J. Saidel, E. Mosovich, J. Biol. Chem. 193 (1951) 397-404. |
| [9] |
A. Patchornik, B. Amit, R.B. Woodward, J. Am. Chem. Soc. 92 (1970) 6333-6335. DOI:10.1021/ja00724a041 |
| [10] |
J.E.T. Corrie, A. Barth, V. Ranjit, et al., J. Am. Chem. Soc. 125 (2003) 8546-8554. DOI:10.1021/ja034354c |
| [11] |
G. Papageorgiou, D.C. Ogden, A. Barth, J.E.T. Corrie, J. Am. Chem. Soc. 121 (1999) 6503-6504. DOI:10.1021/ja990931e |
| [12] |
B. Schade, V. Hagen, R. Schmidt, et al., J. Org. Chem. 64 (1999) 9109-9117. DOI:10.1021/jo9910233 |
| [13] |
J. Hoffmann, U. Kazmaier, Synthesis 47 (2015) 411-420. |
| [14] |
C.P. O'Banion, L.T. Nguyen, Q. Wang, et al., Angew. Chem. Int. Ed. 55 (2016) 950-954. DOI:10.1002/anie.201508767 |
| [15] |
S. Tang, J. Cheng, J. Zheng, Tetrahedron Lett. 56 (2015) 4582-4585. DOI:10.1016/j.tetlet.2015.06.016 |
| [16] |
Y. Shamay, L. Adar, G. Ashkenasy, A. David, Biomaterials 32 (2011) 1377-1386. DOI:10.1016/j.biomaterials.2010.10.029 |
| [17] |
C. Nadler, A. Nadler, C. Hansen, U. Diederichsen, Eur. J. Org. Chem. 14 (2015) 3095-3102. |
| [18] |
M. Mohsen Mahmoodi, D. Abate-Pella, T. Pundsack, et al., J. Am. Chem. Soc. 128 (2016) 5848-8559. |
| [19] |
E. Janett, Y. Bernardinelli, D. Müller, C.G. Bochet, Bioconjugate Chem. 26 (2015) 2408-2418. DOI:10.1021/acs.bioconjchem.5b00473 |
| [20] |
J.S. Wood, M Koszelak, J. Liu, D.S. Lawrence, J. Am. Chem. Soc. 120 (1998) 7145-7146. DOI:10.1021/ja980960+ |
| [21] |
Q. Wang, Z. Dai, S.M. Cahill, M. Blumenstein, D.S. Lawrence, J. Am. Chem. Soc. 128 (2006) 14016-14017. DOI:10.1021/ja065852z |
| [22] |
S. Tang, Z. Wan, Y. Gao, et al., Chem. Sci. 7 (2016) 1891-1895. DOI:10.1039/C5SC03404C |
| [23] |
X. Chen, S. Tang, J. Zheng, et al., Nat. Commun. 6 (2015) 7220. DOI:10.1038/ncomms8220 |
| [24] |
A. Shigenaga, J. Yamamoto, T. Kohiki, T. Inokuma, A. Otaka, J. Pept. Sci. 23 (2017) 505-513. DOI:10.1002/psc.v23.7-8 |
| [25] |
A. Zorzi, K. Devie, C. Heinis, Curr. Opin. Chem. Biol. 38 (2017) 24. DOI:10.1016/j.cbpa.2017.02.006 |
| [26] |
C.J. Whit, A.K. Yudin, Nat. Chem. 3 (2011) 509-524. DOI:10.1038/nchem.1062 |
| [27] |
G.K.T. Nguyen, C.T.T. Wong, J. Biochem. Chem. Sci. 1 (2017) 1-13. |
| [28] |
J.P. Tam, C.T.T. Wong, J. Biol. Chem. 287 (2012) 27020-27025. DOI:10.1074/jbc.R111.323568 |
| [29] |
S.B.H. Kent, Chem. Soc. Rev. 38 (2009) 338-351. DOI:10.1039/B700141J |
| [30] |
G.M. Fang, Y.M. Li, Y.C. Huang, et al., Angew. Chem. Int. Ed. 50 (2011) 7645-7649. DOI:10.1002/anie.201100996 |
| [31] |
J.S. Zheng, S. Tang, Y.K. Qi, Z.P. Wang, L. Lei, Nat. Protoc. 8 (2013) 2483-2495. DOI:10.1038/nprot.2013.152 |
| [32] |
L.Z. Yan, P.E. Dawson, J. Am. Chem. Soc. 12 (2001) 526-533. |
| [33] |
C.T.T. Wong, C.L. Tung, X. Li, Mol. Biosyst. 9 (2013) 826-833. DOI:10.1039/c2mb25437a |
| [34] |
K. Jin, T. Li, H.Y. Chow, H. Liu, X. Li, Angew. Chem. Int. Ed. 56 (2017) 14607-14611. DOI:10.1002/anie.201709097 |
| [35] |
H. Li, S. Dong, Sci. Chin. Chem. 60 (2017) 201-213. |
| [36] |
J. Ma, J. Zeng, Q. Wan, Top. Curr. Chem. 363 (2015) 57-101. |
| [37] |
H. Rohde, O. Seiz, Biopolymers 94 (2010) 551-559. DOI:10.1002/bip.21442 |
| [38] |
J. Yang, J. Zhao, Sci. Chin. Chem. 61 (2018) 97-112. DOI:10.1007/s11426-017-9056-5 |
| [39] |
X. Li, H.Y. Lam, Y. Zhang, C.K. Chan, Org. Lett. 12 (2010) 1724-1727. DOI:10.1021/ol1003109 |
| [40] |
C.L. Lee, X. Li, Sci. Chin. Chem. 59 (2016) 1061-1064. |
| [41] |
Y. Zhang, C. Xi, H.Y. Lam, C.H. Lee, X. Li, Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 6657-6662. DOI:10.1073/pnas.1221012110 |
| [42] |
C.L. Lee, H. Liu, C.T.T. Wong, H.Y. Chow, X. Li, J. Am. Chem. Soc. 138 (2016) 10477-10484. DOI:10.1021/jacs.6b04238 |
| [43] |
T. Kawakami, S. Aimoto, Tetrahedron Lett. 44 (2003) 6059-6061. DOI:10.1016/S0040-4039(03)01463-1 |
| [44] |
R. He, S.H. Chao, Y.J. Tsai, et al., ACS Nano 11 (2017) 6795-6807. DOI:10.1021/acsnano.7b01645 |
| [45] |
M.A. Priestman, D.S. Lawrence, Biochim. Biophys. Acta 1804 (2010) 547-558. DOI:10.1016/j.bbapap.2009.09.005 |
| [46] |
A. Gandioso, M. Cano, A. Massaguer, V. Marchan, J. Org. Chem. 81 (2016) 11556-11564. DOI:10.1021/acs.joc.6b02415 |
| [47] |
Y. Ohmuro-Matsuyama, Y. Tatsu, Angew. Chem. Int. Ed. 47 (2008) 7527-7529. DOI:10.1002/anie.v47:39 |
| [48] |
X. Xie, Y. Yang, Y. Yang, et al., Drug Deliv. 23 (2016) 2445-2456. |
| [49] |
L.T. Nguyen, N.P. Oien, N.L. Allbritton, D.S. Lawrence, Angew. Chem. Int. Ed. 52 (2013) 9936-9939. DOI:10.1002/anie.201305510 |
| [50] |
D.L. Pettit, S.S.H. Wang, K.R. Gee, G.J. Augustine, Neuron 19 (1997) 465-471. DOI:10.1016/S0896-6273(00)80361-X |
| [51] |
K.J. König, J. Microsc. 200 (2000) 83-104. DOI:10.1046/j.1365-2818.2000.00738.x |