 2018, Vol. 29
2018, Vol. 29 



A vast amount of inheritable information is stored in DNA sequences. In eukaryotes, the long informative DNA is packed into a highly compact complex called chromatin. Nucleosomes are the fundamental repeating units of chromatin, which consist of a histone octamer and 1.65 turns of DNA double helix wrapped around (about 147 base pairs). The histone octamer is composed of two copies of each of the core histones H2A, H2B, H3 and H4, the positively charged surface of which makes it a perfect spool for the negatively charged DNA.
Besides serving as the scaffolds for DNA packaging, histones provide an additional level for gene regulation by altering posttranslational modifications (PTMs) on different residues, covering at least 130 unique sites [1-5]. Well-characterized examples of these PTMs include serine phosphorylation, lysine methylation and acetylation [2, 4]. In general, histone PTMs are added and removed by their "writers" and "erasers", respectively. Histone PTMs are considered to regulate DNA-associated processes by two different mechanisms. Firstly, these modifications can directly perturb the histone-histone and DNA-histone interactions to affect the overall structure of chromatin. Secondly, histone PTMs can serve as platform to recruit their recognizing proteins ("readers") to chromatin and execute downstream cellular events [2, 3].
One way to better understand the link between histone PTMs and their cellular functions is to profile the "readers", "writers" and "erasers" of PTMs. Given the fact that most histone PTMs are found on the non-structured tails of histones rather than their globular domains, short synthetic peptides of histone N- or C- termini with less than 40 residues can partly represent PTM state of nucleosomes, making them ideal tools to investigate interactions between histone PTMs and their binding partners. The feasibility of this idea was demonstrated in the 1970s - after Merrifield and coworkers developed solid phase peptide synthesis (SPPS) [6]. In 1974, Allfrey and coworkers showed that histone deacetylases (HDACs) separated from calf thymus could remove acetyl group from lysine-16 of H4 (1–37) but not a shorter synthetic fragment of H4 (15–21) [7]. Five years later, they used a synthetic radioactive K12 and K16 diacetylated H4 (1–37) peptide as substrate of isolated calf thymus HDACs in order to study the mechanism of histone deacetylation [8]. Due to the increasingly more economical synthesis of short peptides, synthetic peptides have become the main tools for identifying histone PTM binding proteins.
In this review we summarize synthetic peptide-based methods that are designed to identify binding partners of histone PTMs. The first part of the review includes the development of peptide pulldown assay, which use rationally designed peptide probes to study histone PTMs. In the second part, peptide-array-based approaches enabling high-throughput screening of PTMs binding partners are discussed. Finally, we briefly discuss the limitations of these peptide-based approaches and describe the construction of nucleosome-based models for investigating histone PTMs-mediated interactions in a nucleosomal context.
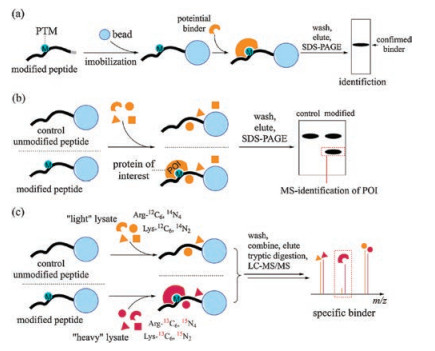
2. Peptide-based pull-down assays to investigate binding proteins of histone PTMsThe method for performing the peptide pull-down assay is described in Fig. 1a. Peptides with a specific tag are first bound onto corresponding resin or beads and treated with protein candidates. After incubation, the beads are washed with a suitable buffer to remove non-specific binding proteins. Finally, the bound proteins are eluted, resolved by SDS-PAGE and identified in different ways.

|
Download:
|
| Fig. 1. Peptide pull-down assays to identify binding proteins of histone PTMs. (a) General workflow of peptide pull-down assays. (b) Unbiased peptide pull-down assay coupled with MS allows identification of unknown POIs. (c) Application of SILAC allows quantitative identification of POIs. POI, protein of interest. SILAC, stable isotope labeling by amino acids in cell culture. | |
{kind=link}
2.1. Development of candidate-based peptide pull-down assays
Early examples that demonstrated the possibility of using the peptide pull-down assay to identify the binding proteins of histone PTMs include the GST pull-down assay with bacterial expressed glutathione S-transferase (GST)-fusion peptides. With GST-fused H2A, H2B, H3 and H4 N-terminal peptides expressed and purified from E. coli, the Grunstein group confirmed that yeast silent information regulators, Sir3 and Sir4, can directly interact with the N-terminal regions of H3 and H4 [9]. Using same peptides, Filetici and co-workers reported that Gcn5 is also a binding protein of H3 and H4 N-termini, and this interaction is through its bromodomain (BRD) [10]. A limitation of GST-fusion peptides is that PTMs on peptides cannot be directly introduced. Required modifications are added through mutagenesis using other canonical amino acids with the same charge and similar backbone length as modified residues to replace unmodified ones. For example, glutamine and aspartate are generally used to mimic acetylation on lysine and phosphorylation on serine, respectively. In 2007, by using expressed GST-H4(1–34) and GST-H4(1–34, K5/8/12/16Q), Côté demonstrated an acetylation-independent interaction between Dot1 and the H4 tail in vitro, which deepens their understanding on how Dot1 localized to nucleosome and methylates K79 on H3 [11].
With the increasing ease and decreasing cost of synthesizing peptides by SPPS, the peptide pull-down assay with synthetic peptides have become accessible for most laboratories, thus accelerating development of the assay and expanding the variety of peptide probes used. In the peptide pull-down assay, the first critical step is to immobilize peptides onto a solid support. Since cysteine is absent at the tail regions of core histones, an early option is to incorporate an additional cysteine at the end of the synthetic peptides, which can be covalently crosslinked onto commercially-available functionalized beads. The Jenuwein group prepared stationary peptide beads by covalently coupling C-terminal cysteine-labeled peptides with POROSTM beads via sulfo-GMBS N-γ-maleimidobutyryl-oxysulfosuccinimide ester), a bifunctional crosslinker. In this way, they prepared immobilized H3 (1–20), H3K9me2 (1–20) and H3S10phos (1–20). These peptide matrices were subsequently used in peptide pull-down assays with several candidates, resulting in the confirmation of HP1 as a binding protein of H3K9me2 [12]. At almost the same time, the Kouzarides group also reported a selective interaction between HP1 chromodomain (CD) and H3 methylated on K9 but not on K4. Similarly, their finding was based on peptide pull-down assays using C-terminal cysteine-labeled H3K4 and H3K9 N-termini peptides with or without methylation, linked to iodoacetylfunctionalized SulfoLink (Thermo) beads [13]. Using the same assay, they reported that H3K4 methylation aborts the interaction between the H3 tail and the nucleosome remodeling and deacetylase (NuRD) complex [14]. Beads activated with iodoacetyl groups were also used in the Reinberg laboratory, where they successfully demonstrated interactions between CD-containing proteins Polycomb(Pc)1, human CHD1 and H3K27me3, H3K4me3 respectively [15, 16].
In addition to chemically crosslinking peptides onto functionalized resin, peptides are more generally conjugated onto avidin beads through the strong interaction between biotin and avidin, which are claimed to have lower backgrounds compared with SulfoLink in peptide pull-down experiments [17]. An early application of biotinylated histone peptides was conducted by Yi Zhang and co-workers. To test their hypothesis that H3K27 methylation is recognized by polycomb (Pc) and facilitates binding of the polycomb response element (PRE), they conjugated biotinylated H3 (19–35) with or without K27 methylation on streptavidin sepharose beads and performed a peptide pull-down assay with 35S-labeled PC or HP1-α separately. By analyzing autoradiographs, they concluded that there was specific binding of Pc to methylated H3K27 [18]. With same design of the peptide pull-down assay but using western blotting as signal outputs rather than autoradiographs, the Grant lab found that the second chromodomain (CD2) of yeast CHD1 bound to the H3K4me2 peptide but not unmodified, acetylated K9 and phosphorylated S10 controls, identifying a new subfamily of CD-containing proteins other than HP1 and the Pc group [19]. A similar assay demonstrated specific binding between Rsc4 tandem BRD region and H3K14Ac [20].
2.2. Identifying protein of interest (POI) from a protein mixtureThe use of autoradiograph and western blotting to obtain signal outputs relies on the knowledge of what candidate proteins should be incubated with immobilized synthetic peptides, which researchers do not always have. To identify an unknown set of binding proteins, peptide pull-down assays should be performed in an unbiased manner. To this end, protein mixtures from lysates are incubated with peptides with or without specific modifications. After SDS-PAGE, the bands that are distinct from the control are analyzed by mass spectrometry (MS) to determine the exact identity of the bands (Fig. 1b). Wysocka et al. performed peptide pull-down assays with biotinylated H3K4me2 and wild-type peptides and nuclear extracts. One single band appears in the K4 di-methylated sample but not the unmodified sample, which was analyzed with MS and identified as WDR5. Following peptide pull-down assays with different biotinylated peptides and GSTfused WDR5 suggested robust and selective binding between methylated H3K4 and WDR5 [21]. Robert et al. treated nuclear extracts with H3K4me3 peptide immobilized on SulfoLink beads. After washing, purified proteins were eluted, resolved by SDSPAGE and then digested with trypsin. Analyzing the sample by MS gave a list of proteins that directly or indirectly bind H3K4me3 [22]. In 2006, several plant homeodomain (PHD)-containing proteins were reported to bind the K4 di- or tri-methylated H3 tail, including BPTF (bromodomain and PHD finger transcription factor) [23, 24] and proteins belong to the ING (inhibitor of growth) family [25, 26]. BPTF was identified by using the biotinylated H3K4me3 peptide as a probe, and unmodified H3, K9me3, K4me2, and S10 P peptides as controls in HKE293 cell nuclear extracts, followed by subsequent analysis with MS [23].
One problem that researchers always encounter with the peptide pull-down assay when the peptide is incubated with lysates is severe background from nonspecific binding proteins. While comparison with the control unmodified peptide sample can rule out the nonspecific binding proteins, it is sometimes ambiguous since it is not a quantitative approach. The development of stable isotope labeling by amino acids in cell culture (SILAC) has provided a quantitative way to identify the bona fide binding proteins among numerous nonspecific ones 27] (Fig. 1c). Application of SILAC in peptide pull-down assays was demonstrated by Vermeulen et al. In their SILAC-assisted peptide pull-down assay using biotinylated H3(1–17) with or without H4me3, they confirmed several known binding partners of H3K4me3, including the above-mentioned CHD1, BPTF and ING family proteins. Novel binding proteins of this modification were also revealed, including all TFIID subunits, PHD finger protein 2 and 8 and Jarid1a. Followup experiments on TAF3 revealed its role as a transcriptional coactivater 28]. In 2010, Vermeulen et al. reported a comprehensive map of histone lysine methylation interactome using the same approach. A number of interactors of H3K4me3, H3K9me3, H3K27me3, H3K36me3 or H4K20me3 were identified at the same time in corresponding peptide pull-down experiments [29].
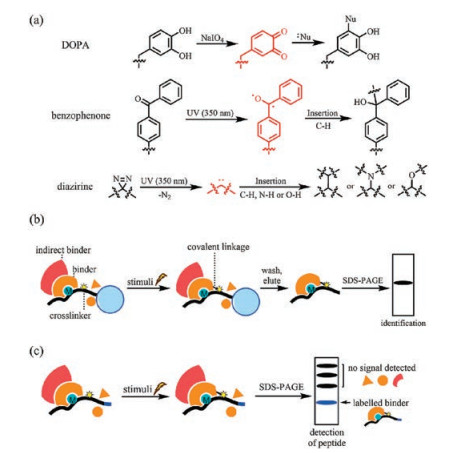
2.3. Capturing weak or transient interactions by crosslinkersAnother drawback of the peptide pull-down assay is the difficulty in finding a suitable wash condition after incubation of immobilized peptides with the protein pool. Due to the noncovalent nature of protein-protein interactions, a milder washing condition usually gives a higher background from nonspecific binding proteins and weak or transient binding proteins can never be detected because even a mild washing condition can abolish these interactions. Harsher washing conditions, however, increases the risk of losing the proteins of interest. Moreover, proteins that bind to the peptide-binding proteins (indirect binders) are usually eluted together with the peptide-binding proteins (direct binders). To differentiate an indirect binder with a direct binder, additional candidate-based peptide pulldown experiments have to be conducted with each binder. To address these problems, crosslinkers are incorporated into synthetic peptides to convert non-covalent interactions between the peptide probe and its binding proteins into covalent ones after incubation with the protein mixture. Crosslinkers are designed to be stable under normal buffer conditions. After exposure to certain stimuli, either chemical or physical (e.g., temperature, light), crosslinkers become activated and link themselves to different components in close proximity, forming strong covalent bonds with binding proteins (Fig. 2a). With covalently-crosslinked peptides and proteins of interest, a strong denaturing buffer can be used in the washing step, leaving only direct binding proteins for identification (Fig. 2b).

|
Download:
|
| Fig. 2. Crosslinkers and general workflows of crosslinking-assisted peptide pull down assay. (a) Structures and mechanisms of commonly used crosslinkers. (b) The covalent bond between peptide probe and binder allows harsh washing conditions, leaving only the direct binders for identification. (c) Additional level of detection provided by peptide probes avoids washing and elution steps when detection is the sole purpose. DOPA, di-ortho-hydroxyphenylalanine. | |
{kind=link}
Cross-linkers not only simplify the choice of washing buffer, but also provide another level of detection. After crosslinking, proteins of interest can also be considered as covalently 'labeled' by the crosslinker containing synthetic peptides. Since SDS-PAGE cannot break this covalent interaction, captured proteins can be detected by the characteristics of our synthetic peptides such as biotin tags, thus avoiding the tedious peptide immobilization and elution steps when detection is the sole purpose (Fig. 2c). The crosslinking properties of DOPA (di-ortho-hydroxyphenylalanine) were first reported by Kodadek and coworkers. Oxidation of DOPA can be selectively achieved using sodium periodate, an inert oxidant to most proteins. After activation by oxidation, an ortho-quinone intermediate is formed and quickly targeted by nucleophiles in spatial proximity, forming the desired covalent linkage (Fig. 2a) [30]. Wen Yuan et al. incorporated DOPA into the N-terminal of Cbiotinylated H3(35–42) (probe 1, Fig. 3a). The peptide was subsequently incubated with the PRC2 complex. After activation by sodium periodate, the complex was resolved and checked for the presence of biotin to see which components were directly crosslinked by the peptide. SUZ12 was recognized as a direct binding partner of H3(35–42) [31].

|
Download:
|
| Fig. 3. Crosslinker-containing probes and their applications. (a) Structures of probes 1–5. (b) An alkyne tag enables CuAAC with azide-tagged fluorophore or biotin after activation of crosslinkers. (c) Schematic diagram illustrating CLASPI and CLASPI-C. CuAAC, copper(Ⅰ)-catalyzed azide-alkyne cycloaddition. CLASPI, cross-linking-assisted and SILAC-based protein identification. CLASPI-C, competition CLASPI. | |
{kind=link}
The presence of crosslinkers also allows the use of bioorthogonal tags in place of the biotin tag. Alkyne is the most commonly used bioorthogonal tag in the peptide pull-down approach. In the presence of alkyne, crosslinked peptides can be conjugated with either azide-tagged fluorophore or azide-tagged biotin through copper(Ⅰ)-catalyzed azide-alkyne cycloaddition (CuAAC) [32]. Conjugated fluorophores allow the presence of binding proteins to be detected with a low detection limit, while conjugated biotin allows pull-down of the binding proteins for subsequent MS analysis (Fig. 3b). This kind of peptide probe was first designed and synthesized by Li et al. Based on the crystal structure of ING2 PHD finger binding to H3K4me3 peptide, they reasoned that A7 at the H3 N-terminal is a suitable residue to be replaced by a crosslinker. An H3K4me3 peptide bearing 4-benzoylphenylalanine (Bpa) in place of A7 and a propargyl glycine (pra) at the C-terminal was synthesized and tested (probe 2, Fig. 3a) [33]. Guided by this work, Yang et al. developed better-performing probes using diazirine as a crosslinker. This kind of probes were demonstrated to be capable of capturing transient interactions between PTM "erasers" and corresponding PTM marks such as Sirt3 and acetylated K9 on H3 (probe 3, Fig. 3a), Sirt5 and malonylated K9 on H3 (probe 4, Fig. 3a) [34]. Using the same principles, probe 5 (Fig. 3a) was designed and synthesized. Using this probe, Sirt2 was identified as a deacylase of myristoylated K9 on H3 [35].
The crosslinking-assisted peptide pull-down assay is more powerful in identifying direct PTM binding proteins in an unbiased way when coupled with the SILAC approach. Cross-linkingassisted and SILAC-based protein identification (CLASPI) was developed by Li et al. in 2012 (Fig. 3c). The interaction between SPIN1 and H3K4me3 was confirmed, while the interaction between the MORC3 zinc finger CW domain and H3K4me3 was identified for the first time [36]. Besides "forward" and "reverse" experiments in traditional SILAC experiments, a competition CLASPI experiment (CLASPI-C) was developed as an affinity filter to identify binding proteins that have a high affinity to PTM marks (Fig. 3c) [37]. Using CLASPI and CLASPI-C, Bao et al. successfully identified Sirt1, Sirt2 and Sirt3 as "erasers" of crotonylation marks on histone H3 [38].
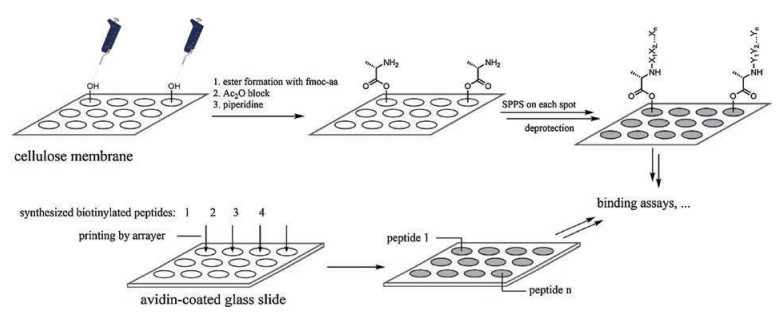
3. High-throughput screening of PTM binding proteins by arraybased approachesSynthetic peptides also contribute to identifying histone PTM binding proteins through array-based experiments. The first peptide array was introduced by Barteling and coworkers in 1984. A total of 208 hexapeptides were synthesized concurrently on packed polyethylene rods, and tested to find out an immunogenic epitope of the immunologically important coat protein of foot-and-mouth disease virus [39]. SPOT synthesis, developed by Frank, allows direct synthesis of peptides on cellulose sheets, which further simplified the preparation of peptide assays and broadened their use (Fig. 4) [40, 41]. However, probably limited by the peptide lengths that can be synthesized by SPOT with satisfactory quality control, SPOT application on histone peptides is less reported. Instead, early application of peptide arrays to identify histone PTM binding proteins relies on conventional SPPS to synthesize the peptides followed by printing them onto glass slides (Fig. 4). In this manner, although greater effort must be made to perform purification of the synthetic peptides, peptide lengths can reach as long as 30–40 residues and the material is usually enough for hundreds of slides [42].

|
Download:
|
| Fig. 4. Schematic diagram illustrating the preparation of peptide arrays by SPOT synthesis (upper) and arrayer printing (lower). | |
{kind=link}
3.1. The peptide microarrays prepared with synthetic peptides
In 2007, the Gozani group prepared a peptide microarray by printing 12 conventionally synthesized biotinylated histone H3 peptides onto streptavidin-coated slides. The prepared slides were incubated with different GST-fused S. cerevisiae PHD fingers, washed, and visualized for the presence of GST to screen the binding proteins of K4 or K36 methylated H3 within the PHD finger-containing proteins in S. cerevisiae. Results demonstrated that most of the candidates did indeed recognize H3K4me3 or H3K36me3 [43]. Using the same approach and greater variation of histone peptides on single peptide microarray slide, RAG2 and ING4 were confirmed as specific binding proteins of H3K4me3 through their PHD domains [44, 45]. In the same laboratory, selective binding between the BAH domain of ORC1 and H4K20me2, but not H4K20me1 or H4K20me3 was demonstrated by peptide microarrays containing 82 distinct histone peptides [46].
Peptide microarrays were also extensively used in the Strahl laboratory. Considering PTMs were increasingly reported to function in a combinatorial way, the biotinylated peptide library that was printed by this group onto slides, in a high-density manner, also included combinatorial PTMs of lysine or arginine methylation, lysine acetylation, threonine or serine phosphorylation besides single PTMs. These peptide arrays demonstrated that binding of BPTF, CHD1 and RAG2 to H3K4me3 was affected by the presence of combinatorial PTMs [47]. By a similar approach, they confirmed that the tandem Tudor domain (TTD) of UHRF1 binds to H3K9me3 in the presence of adjacent H3S10ph, which was previously reported to perturb binding between H3K9me3 and its binding proteins [48]. Follow-up screening by using different mutants of UHRF1 TTD-PHD domains with their arrays containing histone H3 N-terminal tails, however, revealed that binding between H3K9me3 and UHRF1 TTD is less significant than binding between unmodified H3K4 and PHD [49]. Recently, Shanle et al. used their multivalent PTM-containing histone peptide microarray platform to screen over 31 CDs, 39 Tudor or Tudor-like domains as well as several other reader domains (83 in total), and reported the recognition of CBX/HP1 CDs toward H3K23me2/3, Spindlin Tudor domains toward H4K20me3 for the first time [50].
Peptide microarrays can be prepared on diverse surfaces with different coating strategies. A total of 32 biotinylated peptides containing acetylation on known acetylation sites were spotted onto SAM Biotin Capture membrane (Promega) by Zhou and coworkers. Using this membrane, 14 GST-fused BRDs containing yeast proteins were analyzed, and features important for binding between acetylated histones and BRDs were concluded [51]. Zachary et al. immobilized peptides on a gold surface covered with a maleimide-terminated self-assembled monolayer through C-terminal cysteine on the peptides. In this way, the prepared array was readily coupled with MALDI-TOF MS, which they named SAMDI-TOF MS. Using this method, substrates scope of different HDAC activities toward 361 acetylated peptides were profiled [52, 53]. A drawback of the above-mentioned coating methods is that the peptide is immobilized on the plate through certain functional groups or tags, which limits orientation of the peptide, restricting a certain area of the peptide from being recognized by potential binding proteins. To overcome this disadvantage, a functional-group-unbiased photoaffinity approach to conjugate small molecules onto a solid surface was developed by Osada and coworkers [54]. Briefly, a diazirine-containing linker molecule was firstly chemically coupled to a glass slide, followed by printing of small molecules thereon. Under UV radiation, diazirine is activated and covalently captures nearby molecules, in any orientation, to generate an orientation-unbiased array. Inspired by this methodology, Zhao et al. prepared similar kink of arrays with 125 modified histone peptides instead of small molecules. The plate was incubated with eight histone reader proteins and coupled to surface plasmon resonance imaging (SPRi) technology to directly study thermodynamic and kinetic properties between these "readers" and their target peptides. Using this method they developed, MSH6 in Capsella rubella was identified as an H3K4me3 binding partner [55].
3.2. Applications of SPOT-synthesis generated peptide arraysAlthough the synthetic method was developed much earlier, application of SPOT synthesis-generated peptide arrays to investigate binding proteins of histone PTMs was reported much later. It is likely that modern automation of SPOT synthesis by synthesizers broadens its application in this field. An opinion published in 2008 by Arrowsmith and coworkers described three possible applications of SPOT synthesis on epigenetic research, one of which is mapping the specificity of histone binding domains [56]. Peptide arrays generated by automated SPOT synthesis were regularly employed by the Jeltsch group, whose first SPOT synthesized arrays were used to assess the target specificity of histone methyltransferases Dim-5 from N. crassa. Methyltransferase assays were directly conducted on the arrays containing H3(1–21) peptides with different residues mutated to any other amino acids. The substitution of S10 and T11 of H3 by E reduced Dim-5 activity significantly, suggesting that phosphorylation of these two residues may regulate Dim-5 activity [57]. Using similar methods, substrate scopes of methyltransferase G9a, SET7/9 and NSD1 were determined [58-60]. SPOT synthesized arrays were also used to profile sequence specificity of different PTM recognizing domains. ADD domain and PWWP domain of Dnmt3a were shown to specifically recognize unmodified H3K4 and H3K36me3, respectively [61, 62]. In 2012, SPOT synthesized arrays significantly contributed to a landmark study on the human BRD family. Filippakopoulos et al. reported the screening of 33 human BRDs containing proteins over SPOT peptide arrays covering all possible lysine acetylation sites on all four human core histones, dramatically increasing our understanding on this family. New acetylation marks recognized by these proteins were identified while the results showing affinity between BRDs and substrates were generally weak, indicating additional domains may be needed for specific target binding. This group further systematically designed and synthesized a SPOT array containing 11-mer peptides harboring multiple acetylation sites of H3 and H4. Several candidates showed a strong preference toward certain multiply acetylated marks [63].
3.3. Screening against "split-pool" synthesized peptide librariesWhen randomization of several residues in one sequence is required, peptides can be synthesized through the "split-and-pool" approach to generate a larger peptide library. In 1991, the Knapp group and the Cuervo group independently reported synthesis of peptide libraries through the "split-pool" method [64, 65]. Briefly, at the residue where derivation is to be introduced, solid support beads are split equally into different containers. After coupling of different residues in each container, the beads are then combined and mixed thoroughly for subsequent coupling. This process is repeated at every position where derivation of residues is required and, upon completion, each bead will contain only one homogeneous peptide sequence, allowing detection of a "hit" based on the behavior of each bead. The positive beads are subsequently released from the solid support and then sequenced with the aid of MS. Detection of beads with the required sequence was described by Knapp and coworkers in their original work of the "split-pool" strategy [64]. Based on the method they described, Adam et al. developed an "on-bead western" assay to screen positive beads. In brief, the GST-fusion protein of interest was incubated with the library, recognized by a GST-specific primary antibody, followed by incubation with biotinylated secondary antibody and streptavidinconjugated alkaline phosphatase (SAAP). 5-Bromo-4-chloro-3-indolyl phosphate was then added as a substrate of SAAP, the product of which forms a precipitate on the beads, visible as a blue color, assisting the selection of positive beads. With this method, PHD fingers derived from ING2, RAG2, BHC80 and AIRE proteins were screened against a 5000-H3 (1–10) peptide library with randomized PTMs, revealing that binding affinity of these H3K4me3 binding proteins are influenced by neighboring PTMs at R2, T3 and T6, indicating the presence of crosstalk between these marks [66].
3.4. The use of synthetic peptides to screen against protein-domain arraysIn a reversed manner, synthetic peptides carrying specific PTMs can be screened against libraries consist of different protein domains. The protein-domain-array-based approaches were extensively used by Bedford and coworkers [43, 67-70]. The very first protein-domain array chip was generated by spotting GST-fused representative protein domains onto nitrocellulose-coated glass slides, which include members of WW, SH2, SH3, 14-3-3, FHA, PDZ, pH and FF domains. Several biotinylated peptides were conjugated to streptavidin-Cy3 and screened against the array for fluorescence signal. Alternatively, another peptide with a different PTM state was labeled with streptavidin-Cy5 and used at the same time to identify binding preference toward the arrayed domains [67]. This pioneering work demonstrated the feasibility of the protein array approach to investigate binding between different domains and peptides with certain PTMs. This approach was soon employed to identify binding proteins of several lysine methylation marks on histones. The chromatin-associated domain array (CADOR) chip was generated with GST-fused BRD, CD, Tudor, PHD, SANT, SWIRM, MBT, CW and PWWP domains. By incubation of the CADOR chip with different lysine methylated peptides, the authors confirmed several reported interactions and further identified Tudor and MBT domains as new modules that potentially recognize methylated lysine on histone tails [68]. Using the CADOR microarray, TDRD3 was identified as a reader for H4R3me2a and H3R17me2a [69].
4. Limitations and future perspectivesAlthough the use of synthetic peptides has greatly improved our knowledge regarding histone PTMs and their binding partners, the inadequacy of using peptides to recapitulate chromatin PTMs is obvious. The limitation has three aspects; firstly, apart from Nterminal tails, there are increasing numbers of PTMs found on the inner globular domain of core histones, such as H2AR42me1, H2AR88me1, H2BK57me1, H2BR79me1, H3K79ac, H3K79me1/2, H4R55me1, H4K59me1/2 [5]. Recognition of these PTMs tends to rely on tertiary structures of histones and thus limits the use of unstructured synthetic peptides. Secondly, as previously described, increasing evidence has demonstrated multivalent recognition of PTMs at the same time, as well as the presence of crosstalk between PTMs. These can happen for marks spatially close to each other but far away in sequence, or even on different histones [71]. It is obvious that synthetic peptides are not suitable for the applications in these sophisticated cases. Finally, more than one nucleosomes can be involved in the recognition. As an example, modifications on a nucleosome can be "spread" from one to another, leaving little scope for synthetic peptides to study such a phenomenon [72].
One way to overcome these limitations is to generate homogeneously modified histones or histone resembles, reconstitute them into nucleosomes or even nucleosomal arrays and study the complex as an integrated probe for identifying binding proteins. Efforts have already been made to generate these complexes, the key step of which is to generate histones with specific PTMs. Three main strategies have been developed to obtain histones with a certain PTM mark. In a first strategy, by exploiting the fact that cysteine is virtually absent in histones, a single PTM at a specific residue is mimicked by firstly mutating the residue to cysteine and then alkylating the cysteine using a certain reagent [73-76] (Fig. 5). As a second strategy, site-specific genetic incorporation of unnatural amino acids, developed by Schultz and coworkers allows generation of homogeneous histones with specific PTMs [77, 78]. For PTMs present at the histone tails, a semisynthetic approach can be applied. Histone terminal peptides carrying desired PTMs are firstly synthesized by SPPS and subsequently ligated to expressed truncated histones [79, 80]. Nucleosome reconstitution of these modified histones can then be readily achieved by following the report by Luger et al. [81].

|
Download:
|
| Fig. 5. Reactions between cysteine and different alkylation reagents to prepare different PTM mimics. | |
{kind=link}
Compared to these strategies that allow us to obtain sophisticated models, peptide-based approaches are well-developed as well as cost- and time-efficient. Thus, we anticipate that peptide-based approaches will continue to serve as useful tools to preliminarily investigate histone PTMs and their binding partners in the near future.
AcknowledgmentsWe acknowledge support from the Hong Kong Research Grants Council Collaborative Research Fund (CRF No. C7029-15G), the Areas of Excellence Scheme (No. AoE/P-705/16), the General Research Fund (GRF Nos.17125917 and 17303114), the Early Career Scheme (ECS) (HKU No. 709813P), the National Natural Science Foundation of China (Nos. 21572191 and 91753130).
| [1] |
T. Jenuwein, C.D. Allis, Science 293 (2001) 1074-1080. DOI:10.1126/science.1063127 |
| [2] |
T. Kouzarides, Cell 128 (2007) 693-705. DOI:10.1016/j.cell.2007.02.005 |
| [3] |
S.D. Taverna, H. Li, A.J. Ruthenburg, et al., Nat. Struct. Mol. Biol. 14 (2007) 1025-1040. DOI:10.1038/nsmb1338 |
| [4] |
A.J. Bannister, T. Kouzarides, Cell Res. 21 (2011) 381-395. DOI:10.1038/cr.2011.22 |
| [5] |
M. Tan, H. Luo, S. Lee, et al., Cell 146 (2011) 1016-1028. DOI:10.1016/j.cell.2011.08.008 |
| [6] |
R.B. Merrifield, J. Am. Chem. Soc. 85 (1963) 2149-2154. DOI:10.1021/ja00897a025 |
| [7] |
D.E. Krieger, R. Levine, R.B. Merrifield, et al., J. Biol. Chem. 249 (1974) 332-334. |
| [8] |
D.E. Krieger, G. Vidali, B.W. Erickson, et al., Bioorg. Chem. 8 (1979) 409-427. DOI:10.1016/0045-2068(79)90044-0 |
| [9] |
A. Hecht, T. Laroche, S. Strahl-Bolsinger, et al., Cell 80 (1995) 583-592. DOI:10.1016/0092-8674(95)90512-X |
| [10] |
P. Ornaghi, P. Ballario, A.M. Lena, et al., J. Mol. Biol. 287 (1999) 1-7. DOI:10.1006/jmbi.1999.2577 |
| [11] |
M. Altaf, R.T. Utley, N. Lacoste, et al., Mol. Cell 28 (2007) 1002-1014. DOI:10.1016/j.molcel.2007.12.002 |
| [12] |
M. Lachner, D. O'Carroll, S. Rea, et al., Nature 410 (2001) 116-120. DOI:10.1038/35065132 |
| [13] |
A.J. Bannister, P. Zegerman, J.F. Partridge, et al., Nature 410 (2001) 120-124. DOI:10.1038/35065138 |
| [14] |
P. Zegerman, B. Canas, D. Pappin, et al., J. Biol. Chem. 277 (2002) 11621-11624. DOI:10.1074/jbc.C200045200 |
| [15] |
A. Kuzmichev, K. Nishioka, H. Erdjument-Bromage, et al., Genes Dev. 16 (2002) 2893-2905. DOI:10.1101/gad.1035902 |
| [16] |
R.J. Sims Jr, C.F. Chen, H. Santos-Rosa, et al., J. Biol. Chem. 280 (2005) 41789-41792. DOI:10.1074/jbc.C500395200 |
| [17] |
J. Wysocka, Methods 40 (2006) 339-343. DOI:10.1016/j.ymeth.2006.05.028 |
| [18] |
R. Cao, L. Wang, H. Wang, et al., Science 298 (2002) 1039-1043. DOI:10.1126/science.1076997 |
| [19] |
M.G. Pray-Grant, J.A. Daniel, D. Schieltz, et al., Nature 433 (2005) 434-438. DOI:10.1038/nature03242 |
| [20] |
M. Kasten, H. Szerlong, H. Erdjument-Bromage, et al., EMBO J. 23 (2004) 1348-1359. DOI:10.1038/sj.emboj.7600143 |
| [21] |
J. Wysocka, T. Swigut, T.A. Milne, et al., Cell 121 (2005) 859-872. DOI:10.1016/j.cell.2005.03.036 |
| [22] |
R.J. Sims Jr, S. Millhouse, C.F. Chen, et al., Mol. Cell 28 (2007) 665-676. DOI:10.1016/j.molcel.2007.11.010 |
| [23] |
J. Wysocka, T. Swigut, H. Xiao, et al., Nature 442 (2006) 86-90. DOI:10.1038/nature04815 |
| [24] |
H. Li, S. Ilin, W. Wang, et al., Nature 442 (2006) 91-95. DOI:10.1038/nature04802 |
| [25] |
X. Shi, T. Hong, K.L. Walter, et al., Nature 442 (2006) 96-99. DOI:10.1038/nature04835 |
| [26] |
P.V. Pena, F. Davrazou, X. Shi, et al., Nature 442 (2006) 100-103. DOI:10.1038/nature04814 |
| [27] |
S.E. Ong, B. Blagoev, I. Kratchmarova, et al., Mol. Cell Proteom. 1 (2002) 376-386. DOI:10.1074/mcp.M200025-MCP200 |
| [28] |
M. Vermeulen, K.W. Mulder, S. Denissov, et al., Cell 131 (2007) 58-69. DOI:10.1016/j.cell.2007.08.016 |
| [29] |
M. Vermeulen, H.C. Eberl, F. Matarese, et al., Cell 142 (2010) 967-980. DOI:10.1016/j.cell.2010.08.020 |
| [30] |
L. Burdine, T.G. Gillette, H.J. Lin, et al., J. Am. Chem. Soc. 126 (2004) 11442-11443. DOI:10.1021/ja045982c |
| [31] |
W. Yuan, T. Wu, H. Fu, et al., Science 337 (2012) 971-975. DOI:10.1126/science.1225237 |
| [32] |
J.E. Hein, V.V. Fokin, Chem. Soc. Rev. 39 (2010) 1302-1315. DOI:10.1039/b904091a |
| [33] |
X. Li, T.M. Kapoor, J. Am. Chem. Soc. 132 (2010) 2504-2505. DOI:10.1021/ja909741q |
| [34] |
T. Yang, Z. Liu, X.D. Li, Chem. Sci. 6 (2015) 1011-1017. DOI:10.1039/C4SC02328E |
| [35] |
Z. Liu, T. Yang, X. Li, et al., Angew. Chem. Int. Ed. Engl. 54 (2015) 1149-1152. DOI:10.1002/anie.201408763 |
| [36] |
X. Li, E.A. Foley, K.R. Molloy, et al., J. Am. Chem. Soc. 134 (2012) 1982-1985. DOI:10.1021/ja210528v |
| [37] |
X. Li, E.A. Foley, S.A. Kawashima, et al., Protein Sci. 22 (2013) 287-295. DOI:10.1002/pro.2210 |
| [38] |
X. Bao, Y. Wang, X. Li, et al., eLife 3 (2014) e02999. DOI:10.7554/eLife.02999 |
| [39] |
H.M. Geysen, R.H. Meloen, S.J. Barteling, Proc. Natl. Acad. Sci. U. S. A. 81 (1984) 3998-4002. DOI:10.1073/pnas.81.13.3998 |
| [40] |
R. Frank, Tetrahedron 48 (1992) 9217-9232. DOI:10.1016/S0040-4020(01)85612-X |
| [41] |
R. Frank, J. Immunol. Methods 267 (2002) 13-26. DOI:10.1016/S0022-1759(02)00137-0 |
| [42] |
S.B. Rothbart, K. Krajewski, B.D. Strahl, et al., Methods Enzymol. 512 (2012) 107-135. DOI:10.1016/B978-0-12-391940-3.00006-8 |
| [43] |
X. Shi, I. Kachirskaia, K.L. Walter, et al., J. Biol. Chem. 282 (2007) 2450-2455. DOI:10.1074/jbc.C600286200 |
| [44] |
A.G. Matthews, A.J. Kuo, S. Ramon-Maiques, et al., Nature 450 (2007) 1106-1110. DOI:10.1038/nature06431 |
| [45] |
T. Hung, O. Binda, K.S. Champagne, et al., Mol. Cell 33 (2009) 248-256. DOI:10.1016/j.molcel.2008.12.016 |
| [46] |
A.J. Kuo, J. Song, P. Cheung, et al., Nature 484 (2012) 115-119. DOI:10.1038/nature10956 |
| [47] |
S.M. Fuchs, K. Krajewski, R.W. Baker, et al., Curr. Biol. 21 (2011) 53-58. DOI:10.1016/j.cub.2010.11.058 |
| [48] |
S.B. Rothbart, K. Krajewski, N. Nady, et al., Nat. Struct. Mol. Biol. 19 (2012) 1155-1160. DOI:10.1038/nsmb.2391 |
| [49] |
S.B. Rothbart, B.M. Dickson, M.S. Ong, et al., Genes Dev. 27 (2013) 1288-1298. DOI:10.1101/gad.220467.113 |
| [50] |
E.K. Shanle, S.A. Shinsky, J.B. Bridgers, et al., Epigenetics Chromatin 10 (2017) 12. DOI:10.1186/s13072-017-0117-5 |
| [51] |
Q. Zhang, S. Chakravarty, D. Ghersi, et al., PLoS ONE 5 (2010) e8903. DOI:10.1371/journal.pone.0008903 |
| [52] |
M. Mrksich, Acs Nano 2 (2008) 7-18. DOI:10.1021/nn7004156 |
| [53] |
Z.A. Gurard-Levin, J. Kim, M. Mrksich, ChemBioChem 10 (2009) 2159-2161. DOI:10.1002/cbic.v10:13 |
| [54] |
N. Kanoh, S. Kumashiro, S. Simizu, et al., Angew. Chem. 115 (2003) 5742-5745. DOI:10.1002/(ISSN)1521-3757 |
| [55] |
S. Zhao, M. Yang, W. Zhou, et al., Proc. Natl. Acad. Sci. U. S. A. 114 (2017) e7245-e7254. DOI:10.1073/pnas.1704155114 |
| [56] |
N. Nady, J. Min, M.S. Kareta, et al., Trends Biochem. Sci. 33 (2008) 305-313. DOI:10.1016/j.tibs.2008.04.014 |
| [57] |
P. Rathert, X. Zhang, C. Freund, et al., Chem. Biol. 15 (2008) 5-11. DOI:10.1016/j.chembiol.2007.11.013 |
| [58] |
P. Rathert, A. Dhayalan, M. Murakami, et al., Nat. Chem. Biol. 4 (2008) 344-346. DOI:10.1038/nchembio.88 |
| [59] |
A. Dhayalan, S. Kudithipudi, P. Rathert, et al., Chem. Biol. 18 (2011) 111-120. DOI:10.1016/j.chembiol.2010.11.014 |
| [60] |
S. Kudithipudi, C. Lungu, P. Rathert, et al., Chem. Biol. 21 (2014) 226-237. DOI:10.1016/j.chembiol.2013.10.016 |
| [61] |
A. Dhayalan, A. Rajavelu, P. Rathert, et al., J. Biol. Chem. 285 (2010) 26114-26120. DOI:10.1074/jbc.M109.089433 |
| [62] |
Y. Zhang, R. Jurkowska, S. Soeroes, et al., Nucleic Acids Res. 38 (2010) 4246-4253. DOI:10.1093/nar/gkq147 |
| [63] |
P. Filippakopoulos, S. Picaud, M. Mangos, et al., Cell 149 (2012) 214-231. DOI:10.1016/j.cell.2012.02.013 |
| [64] |
K.S. Lam, S.E. Salmon, E.M. Hersh, et al., Nature 354 (1991) 82-84. DOI:10.1038/354082a0 |
| [65] |
R.A. Houghten, C. Pinilla, S.E. Blondelle, et al., Nature 354 (1991) 84-86. DOI:10.1038/354084a0 |
| [66] |
A.L. Garske, S.S. Oliver, E.K. Wagner, et al., Nat. Chem. Biol. 6 (2010) 283-290. DOI:10.1038/nchembio.319 |
| [67] |
A. Espejo, J. Cote, A. Bednarek, et al., Biochem. J. 367 (2002) 697-702. DOI:10.1042/bj20020860 |
| [68] |
J. Kim, J. Daniel, A. Espejo, et al., EMBO Rep. 7 (2006) 397-403. |
| [69] |
Y. Yang, Y. Lu, A. Espejo, et al., Mol. Cell 40 (2010) 1016-1023. DOI:10.1016/j.molcel.2010.11.024 |
| [70] |
Y. Chang, L. Sun, K. Kokura, et al., Nat. Commun. 2 (2011) 533. DOI:10.1038/ncomms1549 |
| [71] |
J.S. Lee, E. Smith, A. Shilatifard, Cell 142 (2010) 682-685. DOI:10.1016/j.cell.2010.08.011 |
| [72] |
P.B. Talbert, S. Henikoff, Nat. Rev. Genet. 7 (2006) 793-803. |
| [73] |
M.D. Simon, F. Chu, L.R. Racki, et al., Cell 128 (2007) 1003-1012. DOI:10.1016/j.cell.2006.12.041 |
| [74] |
F. Li, A. Allahverdi, R. Yang, et al., Angew. Chem. Int. Ed. Engl. 50 (2011) 9611-9614. DOI:10.1002/anie.201103754 |
| [75] |
D.D. Le, A.T. Cortesi, S.A. Myers, et al., J. Am. Chem. Soc. 135 (2013) 2879-2882. DOI:10.1021/ja3108214 |
| [76] |
Y. Jing, Z. Liu, G. Tian, et al., Cell Chem. Biol. 25 (2018) 166-174. DOI:10.1016/j.chembiol.2017.11.005 |
| [77] |
J.W. Chin, Nature 550 (2017) 53-60. DOI:10.1038/nature24031 |
| [78] |
C.J. Noren, S.J. Anthony-Cahill, M.C. Griffith, et al., Science 244 (1989) 182-188. DOI:10.1126/science.2649980 |
| [79] |
M. Holt, T. Muir, Annu. Rev. Biochem. 84 (2015) 265-290. DOI:10.1146/annurev-biochem-060614-034429 |
| [80] |
J.S. Zheng, S. Tang, Y.K. Qi, et al., Nat. Protoc. 8 (2013) 2483-2495. DOI:10.1038/nprot.2013.152 |
| [81] |
K. Luger, T.J. Rechsteiner, T.J. Richmond, Methods Mol. Biol. 119 (1999) 1-16. |