 2018, Vol. 29
2018, Vol. 29 


 ,
Xiao-Xu Chena,
Qian-Qian Yangb,
Liang-Jing Zhub,
Ning-Ning Lib,
Hai-Zhu Yub,
Xiang-Ming Mengb
,
Xiao-Xu Chena,
Qian-Qian Yangb,
Liang-Jing Zhub,
Ning-Ning Lib,
Hai-Zhu Yub,
Xiang-Ming Mengb
b Department of Chemistry, Anhui University, Hefei 230601, China
Conventional peptide therapeutic agents have poor stability in serum and are difficult to be administered orally [1]. Disulfide-rich peptide toxins and their analogs exhibit relatively high stability in vivo and therefore are becoming promising scaffolds for the development of peptide-based drugs in many groups. For example, Craik et al. found a class of disulfide-rich peptides called cyclotides from plants and used them as platforms for the designing peptide drugs for cancer, pain and various other diseases [2]. By screening and engineering conotoxins, Alewood et al. concentrated on screening bioactive peptide molecules that target ion channels, inflammation, GPCR, or growth factor regulators [3]. To accelerate the screening process, recently Lerner et al. constructed an efficient autocrine-based high-throughput combinatorial venom peptide library system [4]. In addition, promising techniques for the screening of disulfide-rich peptidomimetics have also been developed, such as the bicyclic peptide phage screening [5], RaPID system for macrocyclic peptides [6], and the classical combination peptide library [7, 8].
Although the vast majority of pharmacologically active disulfide-rich peptides are still in clinical stage, several peptide drugs of this kind have been recently issued in the market. In 2004, the U.S. Food and Drug Administration approved an atypical analgesic agent for the treatment of severe and chronic pain, Prialt (Fig. 1), which is a synthetic form of the ω-conotoxin peptide [9]. In 2012, linaclotide containing three disulfide bonds was approved by the FDA for the treatment of constipation-predominant irritable bowel syndrome and chronic idiopathic constipation [10]. In 2017, plecanatide containing two disulfide bonds was approved by the FDA for treating chronic idiopathic constipation [11]. The attractive architecture for disulfide-rich peptide toxins is the presence of a rigid conformation that is stabilized by multiple disulfide bonds [12]. The main difficult in synthesizing these toxins is the formation of correct disulfide bonds. There have been a lot of reports on the chemical synthesis and structural engineering of the disulfide-rich peptide toxins. To summarize this type of useful molecule, this paper surveys the discovery and chemical synthesis of disulfide-rich peptide toxins and their analogs.

|
Download:
|
| Fig. 1. Amino acid sequences of plecanatide, prialt and linaclotide, wherein the yellow line represents the disulfide bond | |
2. Discovery
Disulfide-rich peptide toxins can be found in many different organisms, such as rhesus macaques, snakes, spiders, ants, cone snails, scorpions, and also in plants [13]. Normally, they are used by the hosts as chemical weapons to protect against invasive species or to attack preys [14]. Many of them are neurotoxins, acting as antagonists of ion channels, and because of this, they are invaluable molecular tools for studying the biological functions of ion channels [15]. The name of the toxins usually reflects the organisms that the toxins are found. For example, conotoxins refer to the neurotoxic peptides that are found in cone snails; mambalgins are peptide toxins found in venomous black mamba; myrtoxins are peptide toxins found in bull ant; spider toxins are peptide toxins found in spiders; scorpion toxins refer to peptide toxins found in scorpions. The primary structures of peptide toxins are identified mainly by using Edman sequencing and tandem MS, which usually require time-consuming purification steps [16]. In recent years, the structure elucidation of bioactive disulfide-rich peptide toxins has been greatly accelerated by the emergence of advanced analytic technologies, such as electron capture dissociation (ECD) and electron transfer dissociation (ETD) [17]. The development of bioinformatics, such as cDNA library sequencing and proteomic analysis, has greatly accelerated the discovery of disulfide-rich toxins. Several bioinformatics resources centered on peptide toxins have been constructed, such as the ATDB database [18], ClanTox [19], the T3DB database [20], and ConoServer [21]. We anticipate that more bioactive peptide toxins will be found in the near future.
3. Chemical structure and biological functionThe structural feature of the disulfide-rich peptide toxins is the presence of the diverse structural frameworks formed by multiple disulfide bonds. Due to the short length of the amino acid sequence, three-dimensional structures of peptide toxins are mostly analyzed by NMR NOESY/ROESY experiments, and only a little are determined by X-ray crystallography [22]. Here, we briefly introduce the structure and function of several typical disulfiderich peptide toxins (Fig. 2).

|
Download:
|
| Fig. 2. Sequence of six disulfide-rich peptide toxins, wherein the yellow line represents the disulfide bond | |
3.1. Defensins
Defensins are cysteine-rich, positively charged peptides found in vertebrates, invertebrates, and plants [23]. They are used by hosts to resist microbial pathogens and regulate immune responses [24]. There are three types: α-, β-, and θ-defensins. α-Defensins consisted of 29-35 amino acids contain three disulfide bonds with the arrangement of Ⅰ–Ⅵ, Ⅱ–Ⅳ, Ⅲ–Ⅴ[25], while β-defensins consistes of 45 amino acids also contain three disulfide bonds but with the arrangement of Ⅰ–Ⅴ, Ⅱ–Ⅳ, Ⅲ–Ⅵ [26]. Unlike α-, and β-defensins, θ-defensins are backbonecyclized peptides [27]. θ-Defensins are the only known class of cyclic peptides found in mammals, which can be used by cells to prevent HIV-1 infection [28]. In the structure of θ-defensins, three almost parallel disulfide bonds form a specific cyclic cystine ladder architecture [29]. The two constrained β-strand linked by β1-turn are highly constrained by the cystine ladder, inducing θ-defensins to retain their structure at high temperature up to 80 ℃ and maintain their antibacterial activity at physiological salt solution [30]. The chemistry and biology of θ-defensins have been nicely summarized by Craik [13], thus more details on this are not discussed here.
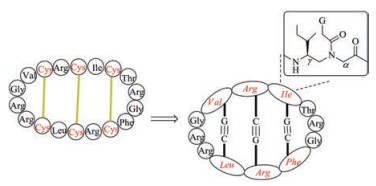
3.2. CyclotidesCyclotides found in plants are characterized by a head-to-tail cyclic structure and a cyclic cystine knot motif (CCK) formed by three disulfide bonds [31]. In the cystine knot, a disulfide bond threads through a ring formed by the other two disulfide bonds. The CCK motif constitutes the core structure of cyclotides, and the side chains of other amino acids are distributed on the surface [32]. Cyclotides can be divided into five loops by six cysteine residues (Fig. 2), of which loop 5 and loop 6 are structurally flexible and thus can be used to graft bioactive amino acid sequence [33-35]. On the basis of the proline configuration in loop 5, cyclotides can be Möbius or bracelet subfamily. The Möbius-type cyclotides have a cis-Pro residue, which forces the peptide bond to twist with a local angle of 180 ℃; the proline in the bracelet-type cyclotides has a trans configuration. A third subfamily of cyclotides is trypsin inhibitor subfamily, with very little sequence similarity to Möbius and bracelet subfamily but also containing a CCK motif [36].
The hydrophobic amino acids on the surface of cyclotides usually contribute to their biological activities. Cyclotides exhibit antimicrobial activity, such as inhibiting the growth and development of Helicoverpa species, Hemochus contortus and Trichostrongylus colubriformis [37-39]. Recent studies showed that cyclotides have ability to resist HIV virus [40]. These bioactivities reported were due to the specific binding of cyclotides to the cell membrane. The hydrophobic amino acids in KB1 can efficiently bind to the phosphatidylethanolamine headgroups on the cell surface, which may protect cells from infection by virus [41]. The cell membrane binding properties also result in the cell membrane permeability of cyclotides. This property has been employed by scientists to graft the bioactive amino acid sequence on the cyclotide scaffolds for the purpose of inhibiting intracellular targets. More details on cyclotides can be found from the review published by Craik [13].
3.3. ConotoxinsConotoxins are a group of neurotoxic peptides containing multiple disulfide bonds, isolated from the venom of genus Conus [42]. The widely studied conotoxins are α-, δ-, κ-, μ-, and ω-conotoxins. α-Conotoxins are small conopeptides with 12–20 amino acids in sequence containing two disulfide bonds, normally inhibit nicotinic acetylcholine receptors (nAChR) [43]. δ-, κ-, and ω-Conotoxins inhibit voltage-dependent sodium channels, potassium channels, and N-type voltage-dependent calcium channels, respectively, and usually contain a cystine knot motif formed by three disulfide bonds [44-46]. μ-Conotoxins, used as inhibitors of voltage-dependent sodium channels, also contain multiple disulfide bonds but lacking a CCK framework [47]. The chemical space of conotoxins is huge, only 0.1% has been investigated. Recently, conotoxin-based drugs have been developed, such as Prialt [48], which is used as a new type of analgesic (a synthetic ω-MVIIA, approved by FDA in 2004).
3.4. BungarotoxinsBungarotoxins are a class of neurotoxic proteins isolated from the venom of kraits, belonging to the three-finger toxin family. There are four types of bungarotoxins: α-, β-, κ-, and γ-subfamily. α-Bungarotoxin comprises 74 amino acids with five disulfide bonds [49]. They can bind to the nAChRs and GABAaR β3 subfamily in synapsis [50]. One remarkable characteristic of α-bungarotoxins is their rigid architecture, which is stable to high temperature and strong acid conditions [51]. Different from α-type, β-bungarotoxin is disulfide-bonded heterodimeric neurotoxin, consisting of a functional PLA2 domain and a Kunitz-type domain, which are linked by an inter-strand disulfide bridge [52]. The binding of β-bungarotoxin to the presynaptic terminal can block the release of acetylcholine and induce apoptotic death of human neuroblastoma SK-N-SH cells [53]. γ-Bungarotoxin is composed of 68 amino acids and five disulfide bonds [54]. The last bungarotoxin is κ-subfamily. κ-Bungarotoxin comprises 66 amino acids with a antiparallel β-sheet structure stabilized by five conserved disulfide bonds [55]. Mambalgins and mambaquaretins, recently isolated from the venom of black and green mamba, similar to bungarotoxins, also belong to three-finger toxin family [56]. Intriguingly, mambalgins do not induce motor dysfunction, convulsions, apathy, flaccid paralysis or death to mice after central injections, but instead produce analgesic effects against neuropathic pain [57]. Besides, Bougis et al. found two neurotoxins, MmTX1 and MmTX2 in the venom of coral snake, with three-demensional structure similar to γ-bungarotoxin [58].
3.5. Scorpion toxinMost neurotoxins isolated from the venom of scorpions can block neuronal transmission by targeting potassium or sodium channels [59]. So far, around 730 scorpion toxins have been found from 56 species. The topological structure of the scorpion toxins can be divided into four types. The most common core topology is cystine-stablized α/β (CS α/β) motif in which an α-helix and a β-sheet are covalently connected by two or multiple disulfide bonds [60]. The second topology is cystine-stablized α-helix-loophelix (CS α/α), where two short α-helices are connected by a β-turn [61]. The third is the well-known topological structure: cystine knot motif. The recently discovered fourth topology is disulfide-directed β-hairpin, which is regarded as the ancestral fold of cystine knot [62]. Due to the limited amount of pure samples by purification and chemical synthesis, only a few has been clarified in biological function. With the advent of advanced purification and synthesis techniques, there will be more interesting discoveries on scorpion toxins.
3.6. Spider toxinsMore than 40, 000 different species of spiders exist on Earth, and each single venom may contain up to 1000 peptides, thus conservatively estimating that 10 million bioactive peptides are present in spider venom [63]. The library of spider toxins is huge, but currently only less than 0.01% has been determined in structure and bioactivity. Spider toxins are usually antagonists of Nav channel proteins. NaSpTxs isolated from spider venom are capable of targeting Navs channel proteins and are used as skeletons for developing analgesic drugs targeting Nav. NaSpTxs contains a wellknown topology named inhibitor cystine knot, and therefore are resistant to high temperature and enzyme degradation [64]. A relatively comprehensive studied NaSpTxs is protoxin Ⅱ, containing 30 amino acids and 3 pairs of disulfide bonds, which can be used to effectively block hNav1.7 [65]. It has recently been found that Hm1a and Hm1b can selectively activate Nav1.1, and promote a deeper understanding of the role of Nav1.1 in pain mechanisms [66]. Spider venom also contain peptide toxins that target Kv channel protein. For example, Hanatoxins containing 35 amino acids and 3 pairs of disulfide bonds, can effectively target Kv2.1 and Kv4.2 [67]. Besides, Julius recently found a rare bivalent peptide toxin in Tarantula, which can activate the function of capsaicin receptor [68].
3.7. Other toxinsDisulfide-rich peptide toxins are also found in other organisms. OvCNP found in duck-billed platypus can cause the influx of Ca2+ in Neuroblastoma cells [69]. μ-SLPTX-Ssm6a, found in centipede venom, contains 46 amino acids and a CCK structure formed by 3 pairs of disulfide bonds [70]. μ-SLPTX-Ssm6a, having analgesic effect more than morphine in the rodent model, can selectively inhibit Nav1.7 channel toxin. SSD609 containing 47 amino acids, isolated from centipede Scolopendra subspinipes dehaani (SSD), can inhibit Iks channel by interacting with the KCNE1 auxiliary subunit [71, 72]. Anthopleurin B, found in the sea anemone venoms, is capable of suppressing the function of Navs at nanomolar concentrations [73]. Mp1a, found in Myrmecia pilosula, is a heterodimeric peptide containing 49 amino acids and was reported to have antimicrobial, membrane-disrupting and nociceptive activities [74]. Apamin, an 18 amino acid peptide containing a pair of two pairs of disulfide bonds, found in bee venom, was reported to be able to selectively block SK channel [75].
4. Formation of correct disulfide bond architectureOnly a limited amount of pure peptide neurotoxins can be isolated from the venom, thus hampering the structure-activity relationship studies. Two methods can be employed to produce pure substances. The first is the use of recombinant cell technology to transform E. coli or yeast to express peptide neurotoxins [76]. Due to the inherent toxicity, usually, the level of toxin expression is not high. In addition, the introduction of unnatural amino acids is challenging for recombinant cellular systems. Taking into account the small size of peptide toxins, many laboratories prefer to use chemical synthesis method. The linear polypeptide sequence of peptide toxins can be readily synthesized by solid phase peptide synthesis or peptide fragments ligation based on native chemical ligation [77-91]. The key to chemical synthesis of peptide toxins is the formation of correct disulfide bonds. The formation of correct disulfide-bond framework can be performed by using oxidative folding strategies or using orthogonal cysteine-protecting groups.
4.1. Oxidative folding to form disulfide bonds in one stepProtein folding is sensitive to a multitude of factors such as pH value, ion concentration, environment temperature. In a cellular environment, a set of complex protein machinery is required for the folding of linear polypeptides translated from mRNA [92]. It contains several molecular chaperones, such as GroEL for the prevention of the aggregation of protein molecules and peptidylprolyl cis-trans isomerases for catalyzing peptideyl-proline bond isomerization. The folding of proteins usually follows Anfinsen's rules [93]. That is, the native structure of a protein depends on the primary amino acid sequence of the protein. Since non-native disulfide bonds may lead to misfolding, the folding of disulfidecontaining peptide toxin is more complex than the disulfide-free peptide, requiring protein disulfide isomerase to rearrange the incorrectly formed disulfide bonds. Several classic studies indicated that the correct formation of the disulfide bond is driven by protein folding [94-96]. In other words, intramolecular disulfide bond is actually employed by Nature as an additional element for stabilizing the 3D architecture of protein. Using an immobilized molecular chaperone system, Fersht et al. achieved the refolding of denatured scorpion toxin Cn5 with a recovery yield of 87% [92].
The formation of correct disulfide bond can also be conducted in vitro by using conditions that mimic physiological environments (Table 1). The commonly used buffer is a slightly alkaline aqueous buffer (pH 7.5–8.5). In order to prevent peptide aggregation, 0.1– 1.5 mol/L guanidine hydrochloride or 0.5–3.0 mol/L urea are often added. The reaction of forming correct disulfide bond is usually carried out under GSH/GSSG/peptide conditions (e.g., 100:10:1). DMSO can also be used as an oxidative reagent in this process. Although a large number of disulfide-rich peptide toxins can fold correctly in vitro, the in vitro strategy has several inherent drawbacks. First, the folding yield and the framework of disulfide-bond linkage are highly dependent on their amino-acid sequence. The folding of non-naturally modified disulfide-rich peptide toxin is more difficult than its unmodified form. Second, since the kinetics of oxidative folding depends on several parameters, the folding conditions need to be optimized, such as the use of different buffers, altering the oxidant or changing the polypeptide concentrations.
|
|
Table 1 Common conditions used for oxidative folding of Cys-rich peptides |
{kind=link}
{kind=link}
There are special conditions for the folding of disulfide-rich peptide toxins. Durek et al. found that the folding of the synthetic linear polypeptide mambalgin-2 was efficient under conventional conditions (100 mmol/L Tris, 500 mmol/L Gn ·HCl, 8 mmol/L GSH, 1 mmol/L GSSG, pH 8.0) [97]. In the chemical synthesis of Ts1 toxin, Kent et al. showed that the concentration of the polypeptide and the incubation temperature are important for Ts1 folding [98]. The native and bioactive isomer Ts1 cannot be formed under the following conditions: polypeptide 0.5 mg/mL, 100 mmol/L Tris, 0.5 mol/L Gn·HCl, 8 mmol/L cysteine, 1 mmol/L cystine, room temperature. However, by lowering the temperature to 4 ℃ and peptide concentration to 0.01 mg/mL, the folding of Ts1 becomes efficient. In the process of synthesizing another peptide toxin-Ts3, Kent et al. encountered the difficulty of Ts3 folding [99]. After carefully screening, bioactive isomer of Ts3 was obtained under very complex conditions: 0.02 mg/mL polypeptide, 2.0 mmol/L GSH/1.0 mmol/L GSSG, 0.5 mol/L L-arginine·HCl, 1.0 mmol/L EDTA, 0.1 mol/L Tris, 20% DMSO, pH 8.5, room temperature. In another project, Kent et al. found ShK toxin can readily fold into the native 3D architecture by air oxidation in 50 mmol/L NH4OAc (pH 8.0) [100]. Prior to the experiments, it is difficult to anticipate the appropriate conditions for toxin folding.
4.2. Regioselective disulfide-bond formation via orthgonal thiolprotecting groupsThe small difference in the thermodynamic stability between the natural conformation and non-natural conformation of a disulfide-rich peptide toxin sometimes leads to failure of its folding by in vitro oxidative strategies [101]. In this case, it is necessary to adopt chemical strategies for regioselective disulfidebond formation. The key to this approach is the choice of orthogonal thiol-protecting groups [102].
According to the conditions used for the liberation of thiol group of Cys, thiol-protecting groups used in solid phase peptide synthesis can be divided into the following categories: acid-labile, alkali-labile, heavy metal ion-labile, reducing reagent-labile and hydrazine-labile protecting groups. The acid-labile protecting groups are widely used in SPPS, including Mob, Meb, Trt, Dpm, Mmt. Mob and Meb are used in Boc-based SPPS, requiring strong acids such as HF or TFMSA to deprotect. Trt is commonly used in Fmoc-based SPPS, requiring at least 25% TFA to release the sulfhydryl group. In many cases, part of the Trt groups is removed even by 1% TFA. Dpm, which can be only cleaved under over 60% TFA, can be used to replace Trt to avert the undesired deprotecting issue [103]. Mmt is ultra sensitive to TFA, requiring only 1% TFA for deprotection. Recently, Liu et al. invented a unique type of acidlabile Cys protecting group: Hmb [104]. The removal of Hmb consists of two steps. In the first step, Hmboff was converted into Hmbon by releasing phenolic hydroxyl group through cyclizationelimination reaction under neutral aqueous solution. Subsequently, Hmbon was removed by concentrated TFA. Alkaline-labile protecting groups contain Fm and Tfacm. Fm is unstable to piperidine, and therefore usually is used in Boc-based SPPS. The latter one Tfacm-protected Cys can be efficiently converted into Cys by hydrolysis at pH 11 while requiring TCEP in order to prevent the base-initiated elimination of disulfide-formed Cys [84]. The representatives of heavy metal ion-labile protecting groups are Acm and Phacm, which can be removed by the toxic reagents, such as AgOAc or Hg(OAc)2 [105]. It is noteworthy that Phacm can be quickly enzymolysis by Penicillin G Acylase [106].
Remarkably, Ashraf et al. recently found that Acm and Phacm can also be efficiently deprotected by a relatively low toxic palladium reagent [107-109]. The reducing reagent-labile protecting groups are StBu, STmp, SNpys, pNB, Pac, Pocam and Msbh. StBu can be removed rapidly by TCEP but sometimes taking several hours when using thiols as reducing reagents. By contrast, STmp and SNpys are very sensitive to thiols and can be removed by DTT in less than 10 min [110]. The deprotection of PNB, Pac and Pocam requires Zn/AcOH to reduce nitro to amino group or keto to alcohol group [111]. Msbh, recently developed by Alewood, is removed by conversion of sulfoxide into thioether by DMS/TFA [112]. The hydrazine-labile protecting group is Hqm, which can be removed by 5% hydrazine [113]. More details on Cys-protecting groups can be found from the review published by Albericio [101].
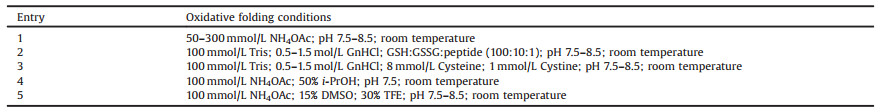
The regioselective construction of disulfide bonds mainly relies on the use of orthogonal thiol-protecting groups. The step for the formation of disulfide bonds can be carried out on solid phase or in solution. To illustrate this process, we show the chemical synthesis of human hepcidin, which contains four pairs of disulfide bonds [114]. Eight cysteine residues in the primary sequence of human hepcidin are protected by four different kinds of protecting groups, namely Trt, Acm, Meb and Msbh (Fig. 3). After completing Fmocbased SPPS, the linear polypeptide was released from the resin by TFA, in this step the Trt groups on Cys13 and Cys16 were also removed. The formation of disulfide bond between Cys13 and Cys16 was accomplished by DMSO-mediated oxidation. In the second step, the removal of Acm and the corresponding disulfidebond formation were accomplished in one step with the aid of iodine under weak acid condition. Afterward, Meb-protected Cys3 and Cys19 were converted into unprotected Cys by HF and oxidized to the disulfide form by iodine. Finally, Msbh-protected Cys4 and Cys12 were removed and transformed to disulfide bond by NH4I/ DMS/TFA. Although the orthogonal thiol-protecting group can guide the regioselective formation of the disulfide bond, many steps are involved in this strategy, therefore usually the final yields are low.

|
Download:
|
| Fig. 3. Chemical synthesis of hepcidin via orthogonal thiol-protecting groups | |
{kind=link}
5. Chemical synthesis of disulfide-rich peptide toxin analogs
The instability of disulfide bond to reductive physiological environments to some extent impedes the application of these kinds of peptides as therapeutic leads. To this end, many novel structures that mimic the disulfide bond have been increasingly developed. Disulfide-rich peptide toxin analogs can maintain the biological function of native peptide toxins, thus catching the interest of many pharmaceutic companies. The following describes the main structure used to mimic the disulfide bridge.
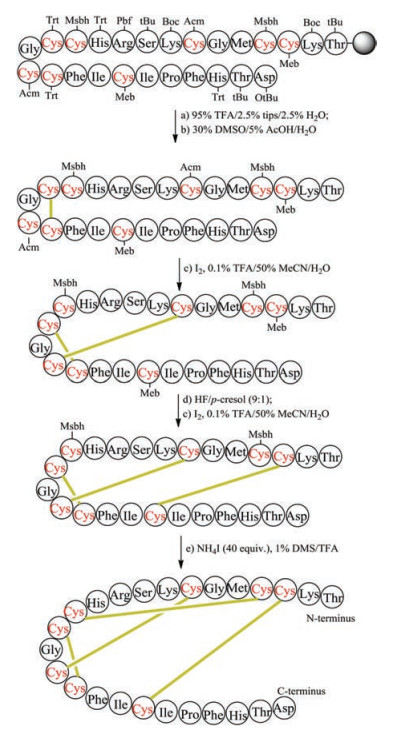
5.1. Unsaturated dicarba bridgeChemical synthesis of unsaturated dicarba mimics was achieved by replacing two cysteine residues at specific positions with L-allylglycine. After the completion of precursor linear peptide by Fmoc-based solid peptide synthesis, ring-closing metathesis was conducted to generate an unsaturated dicarba bridge (Fig. 4). As expected, the unsaturated dicarba bridge affords two geometric isomers, named as cis- and trans-stereochemistry. To define the biologically functional isomer, it is necessary to purify the mixture by RP-HPLC and analyze their corresponding 3-D structure by NMR. Using this strategy, Vederas prepared dicarba analogues of peptide hormone oxytocin, and found the biological activity of cis compound is much higher than the trans compound [115]. Interestingly, during the study of antimicrobial leucocin analogs, Vederas observed that the precursor linear peptide containing allylglycine residues but without ring formation has biological activity similar to the nature parent leucocin, presumably caused by the unexpectedly noncovalent interactions of ally glycine residues [116]. The work on dicarba alfa-conotoxins done by Robinson demonstrated that the dicarba-bridge can be used to improve their receptor subtype selectivity [117, 118].

|
Download:
|
| Fig. 4. Chemical synthesis of unsaturated dicarba-leucocin | |
{kind=link}
5.2. Saturated dicarba bridge
Owing to the lack of cis and trans isomers, the purification of saturated dicarba mimics is much easier than unsaturated dicarba mimics. Two approaches can be used to prepare saturated dicarba mimics. In the first method, olefin metathesis is used to cyclize the linear precursor, followed by reducing olefin using 2, 4, 6- triisopropylbenzenesulfonyl hydrazide (TPSH) [119, 120]. The advantage of this method is that the used L-allylglycine is commercially available, thus one can easily perform this step (Fig. 5). The disadvantage of this method is that only one disulfide bond can be replaced by this strategy. The second method is the use of diaminodiacid-based solid-phase synthesis. In this method, orthogonally protected diaminodiacids compatible with FmocSPPS are required to be prepared first. The biscarba diaminodiacids can be synthesized by Kolbe electrolytic decarboxylative crossdimerization. To avoid interference with Fmoc chemistry, allyl/ allyloxycarbonyl(Alloc) or p-nitrobenzyl (pNb)/p-nitrobenzyloxycarbonyl paired groups are taken to protect a amino and a carboxyl group. By using two sets of protected diaminodiacids, Liu et al. successfully synthesized biscarba analogues of tachyplesin I (TPI- 1), which showed higher serum stability than native TPl-1 that contains two native disulfide bonds [121]. Recently, Liu et al. found the replacement of one disulfide bond with dicarba bridge can facilitate the folding of disulfide-rich peptides, such as μ-conotoxin SIIIA, hepcidin and hepcidin-1 [122-126].

|
Download:
|
| Fig. 5. Chemical synthesis of saturated carba bridge-containing peptide | |
{kind=link}
5.3. Thioether bridge
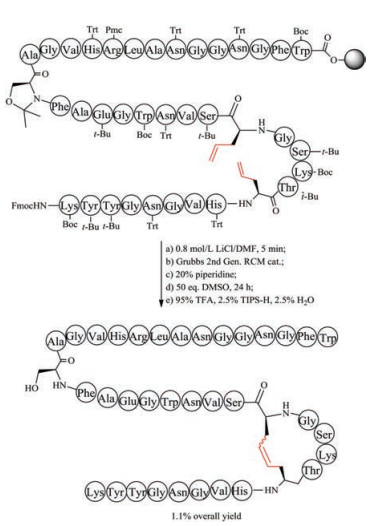
Thioether-crossed-linked amino acids are quite common in lantipeptides. In organisms, the formation of thioether-containing peptides normally involves two biological steps, including serine/ threonine dehydration performed by dehydratases, and thioether cyclization performed by cyclases [127, 128]. Thioether bridge has been used by chemists to replace disulfide bond of peptide toxins, and was regarded as an effective method to retain biological function of native precursors as this replacement only result in a minimal structural perturbation compared with other mimics. The first method to form thioether bridge was achieved by S-alkylation of cysteine with bromoacid or by thiol-ene reaction to crosslink two cysteine residues directly during solid phase peptide synthesis and has been applied to the synthesis of thioether analogs of conotoxin G1 and oxytocin [129-131]. However, owing to the instability to piperidine, bromoacid requires to be coupled to peptide at the final step, and thus this route is not suitable for replacement of more than two disulfide bonds. Interestingly, this strategy has been used in expanded code system to improve the stability of protein. An alternative approach involved the use of orthogonally protected cystathionine-containing diaminodiacids as building blocks, followed by Fmoc-SPPS and cyclization (Fig. 6). This building block approach can be used to place thioether bridge at any position with complete positional freedom. The preparation of cystathionine-containing diaminodiacids is easy and effective, because the reaction only involved switching protecting groups and the conversion of functional groups.

|
Download:
|
| Fig. 6. Chemical synthesis of thioether bridge-containing compstatin | |
{kind=link}
Using two enantiomerically pure, orthogonally protected cystathionine building blocks, Lambris et al. synthesized several thioether bridge-containing compstatin analogues, and found these analogues maintained their high binding affinity for their biological target, comparable to the original disulfide-bridged peptides [132]. Compared with cystathionine bridge, lanthionine linkage is one bond shorter and may be different in conformational preferences [133]. Recently, Tabor et al. successfully synthesized several lanthionine-bridged analogues of ProTx-Ⅱ by using (2R, 6R)-(allyl, Aloc/Fmoc)-lanthionine, and investigated their folding under different oxidation conditions, and reported the first X-ray crystal structure of spider venom peptide not bound by a substrate [134]. Besides, Cramer et al. described a simple protocol to convert disulfide bridges into stable methylene thioacetals by using CH2I2 in the presence of Et3N. The protocol has been used in the synthesis of a variety of biologically functional cyclic bioactive peptides, such as octreotide, oxytocin, vasopression, somatostatin, bactenecin and α-conotoxin MII in high yields [135].
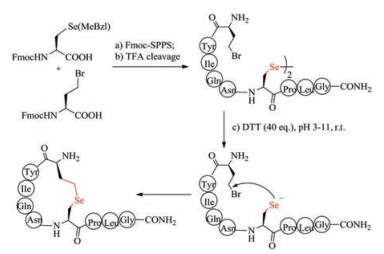
5.4. Selenoether bridgeS-Alkylation of cysteine used in the formation of thiolether bridges is often sluggish, and therefore has not been widely used. Selenocysteine has biochemical property almost similar to cysteine, and thus is regarded as a perfect substitution for cysteine [136]. Despite structural similarity to cysteine, selenocysteine (Sec) exhibits higher nucleophilicity than cysteine, and thus can react easily with bromoacid to form selenoether (Fig. 7). Under pH 5-11, the intramolecular reaction between selenocysteine and γ-bromo-homoalanine finished almost quantitatively within 2 h, whereas the time for S-alkylation took more than 5 h with relatively low yields. Non-reducible selenoether oxytocin was readily prepared by Sec-alkylation and was tested to inhibit colonic nociceptors both in vitro and in vivo comparable to native oxytocin [137]. Selenolanthionine bridge is one bond shorter than selenocystathionine bridge. Alewood et al. recently synthesized lanthionine-like cyclic peptide by substituting cysteine with selenocysteine residue [138]. The simplicity of selenoether bridge formation highlights their promise in the preparation of disulfide bond mimics.

|
Download:
|
| Fig. 7. Chemical synthesis of selenoether bridge-containing oxytocin | |
{kind=link}
5.5. Diselenide bridges
When peptide toxins comprise more than two pairs of disulfide bonds, laborious work on introducing orthogonal thiol-protecting groups is required to achieve regioselective disulfide bond formation. The much higher acidity of Sec than Cys results in the preferential formation of diselenide bond over disulfide bond. The replacement of two Cys residues with Sec was reported to be able to direct the correct folding of α-conotoxin and μO-conotoxin MrVIB, avoiding the formation of undesired disulfide isomers [139]. Owing to the lack of sulfur isotope, disulfide bond localization is poor by NMR. In contrast, diselenide bridge can be visualized directly by 77Se NMR spectroscopy [140]. In a model system, Alewood et al. determined 3D structure of [Sec13, Sec14] analogue of spider toxin κ-ACTX-Hv1c directly by 77Se NMR spectroscopy [141]. Although appealing property owned by Sec, its incorporation by Fmoc-chemistry usually encounters the problem of the racemization and β-elimination of Sec.
5.6. Lactam bridgeLactam bridge can also be used to replace disulfide bond to stabilize the secondary structure of peptides [142-146]. In this strategy, during Fmoc-SPPS, the target disulfide bond-associated cysteine residues are substituted by Glu and Lys with side chains protected respectively with ally ester and allyl urethane (Fig. 8). After the accomplishment of peptide assembly, palladium reagent is used to selectively deprotect ally ester and ally urethane. Then, the cyclization of Glu and Lys is carried out with HBTU-NMM or PyBOP-NMM. Barany et al. synthesized several lactam analogues of α-conotoxin SI and found most of them can tightly bind to nicotinic acetylcholine receptors, albeit with a relatively low affinity [147].Of note, (i, i + 4) lactam bridge has been used as a successful approach to stabilize α-helices. Using this protocol, Norton et al. designed and synthesized truncated analogues of μ-KIIIA that contain a lactam bridge at different positions [148].

|
Download:
|
| Fig. 8. Chemical synthesis of lactam bridge-containing α-conotoxin SI. | |
{kind=link}
5.7. Triazole bridges
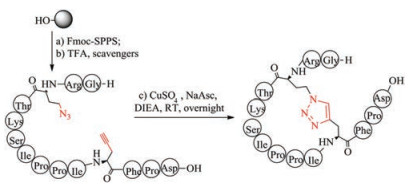
Triazoles exhibit excellent chemical stability to proteases, isomerases and reducing agents and are therefore used as an attractive alternative to disulfide bridges. The formation of triazole bridges is based on click chemistry between the azide functionalized and alkynyl functionalized amino acids (Fig. 9). Depending on the type of catalyst used in the reaction, the triazole bridges can be 1, 4-disubstituted 1, 2, 3-triazole, using copper (Ⅰ) as a catalyst, and 1, 5-disubstituted 1, 2, 3-triazoles, using ruthenium (Ⅱ)-based catalysts [149-152]. Both structures prove to be useful disulfide mimics that allow the conformation of the amide backbone to be fixed. Tachyplesin-Ⅰ (TP-1) analogs with two disulfide bonds replaced by 1, 4-disubstituted-1, 2, 3-triazoles showed similar tertiary structure and biological activity to wild-type disulfide bridged TP-Ⅰ [153]. Conotoxin MrIA mimetic, in which one disulfide bond is replaced by a 1, 4-disubstituted 1, 2, 3-triazole bridge, also retains the original three-dimensional structure, and inhibits the function of noradrenaline transporter in vivo and in vitro [154]. In another paper, replacing the disulfide bond of sunflower trypsin inhibotor-I (SFTI-1) with 1, 5-disubstituted 1, 2, 3-triazole did not disrupt the 3D structure and also the inhibitory activity of SFTI-I [155]. To avoid the potential synthesis difficulties associated with the post-chain assembly of 1, 2, 3-triazole, Hu et al. chemically synthesized diaminodiacids that contain two species of the 1, 2, 3- triazoles group and used them as building blocks to simplify the construction of biologically active thanatin derivatives [124].

|
Download:
|
| Fig. 9. Chemical synthesis of thiazole bridge-containing sunflower trypsin inhibitor-1 | |
{kind=link}
5.8. Arylation of cysteine
It has been demonstrated that perfluorinated aromatics can selectively react with cysteine in the presence of other nucleophilic amino acids such as lysine, histidine, tryptophan and tyrosine [156]. This chemistry has been used to lock unprotected peptides into the α-helical conformation. Recently, Wu et al. found that the reactivity of 2, 3, 5, 6-tetrafluoroterephthalonitrile (4F-2CN) to cysteine is higher than that to penicillamine (Fig. 10). By substituting glycine and the second cysteine residue of linear sunflower trypsin inhibitor-1 with penicillamine, they prepared a bioactive tricyclic SFTI-1 by arylation of cysteine with 4F-2CN [157-160].

|
Download:
|
| Fig. 10. Chemical synthesis of 4F-2CN-cyclized sunflower trypsin inhibitor-1 | |
{kind=link}
5.9. Noncovalent Watson-Crick hydrogen bonds
The abovementioned methods used to displace disulfide bonds are based on the formation of covalent bonds. The distinct cyclic cystine ladder architecture shown by θ-defensins provides an excellent example to mimic disulfide bonds by noncovalent Watson-Crick hydrogen bonds (Fig. 11). By using γ-backbone peptide nucleic acids containing the original chemical functionalities of native RTD-1 to substitute six cysteine residues, Ly et al. designed a cyclic RTD-1 mimic showing antimicrobial activities as the wild type one [161].

|
Download:
|
| Fig. 11. θ-Defensin mimic containing γPNA scaffold | |
{kind=link}
6. Conclusion and outlook
Herein, we have summarized recent advances on cysteine-rich peptide toxins including their discovery, structure elucidation, chemical synthesis and structure engineering. The diversity of the three-dimensional structure and the variability of the amino acid sequence confer a variety of bioactivity to the disulfide-rich peptide toxins. The combination of chemically structural engineering and directed evolution techniques has led to the discovery of a large number of bioactive peptide candidates, and many of them are being at the clinical stage. Using bacterial display library and C-to-N cyclization strategy, Daugherty et al. identified a novel cyclotide that can inhibit the migration of endothelial cell by selectively targeting to Neuropilin-1 and 2 [162]. By positional scanning an α-conotoxin synthetic combinatorial library, Armishaw discovered a potent and selective α3β4 nAChR antagonist, TP-2212-59 [163]. In addition, Goldstein et al. constructed a phage-display library using sea anemone toxin ShK as a framework and identified several peptide toxin blockers of KcsA potassium channel [164]. With the emergence of high quality peptide toxin libraries and highly efficient synthesis methods [165-168], more pharmaceutic-related peptides will be discovered and investigated [169, 170].
AcknowledgmentThis work was supported by National Natural Science Foundation of China (No. 21778001).
| [1] |
K. Fosgerau, T. Hoffamnn, Drug Discov. Today 20 (2015) 122-128. DOI:10.1016/j.drudis.2014.10.003 |
| [2] |
B.B. Carstens, G. Berecki, J.T. Daniel, et al., Angew. Chem. Int. Ed. 55 (2016) 4692-4696. DOI:10.1002/anie.201600297 |
| [3] |
T. Durek, I. Vetter, C.I.A. Wang, et al., ACS Chem. Biol. 8 (2013) 1215-1222. DOI:10.1021/cb400012k |
| [4] |
H. Zhang, M. Du, J. Xie, et al., Angew. Chem. Int. Ed. 55 (2016) 9306-9310. DOI:10.1002/anie.201603052 |
| [5] |
C. Heinis, T. Rutherford, S. Freund, G. Winter, Nat. Chem. Biol. 5 (2009) 502-507. DOI:10.1038/nchembio.184 |
| [6] |
T. Passioura, T. Katoh, Y. Goto, H. Suga, Annu. Rev. Biochem. 83 (2014) 727-752. DOI:10.1146/annurev-biochem-060713-035456 |
| [7] |
Y. Li, A. Gould, T. Aboye, et al., J. Med. Chem. 60 (2017) 1916-1927. DOI:10.1021/acs.jmedchem.6b01689 |
| [8] |
K. Jagadish, A. Gould, R. Borra, et al., Angew. Chem. Int. Ed. 54 (2015) 8390-8394. DOI:10.1002/anie.201501186 |
| [9] |
J.G. McGivern, Neuropsychiatr. Dis. Treat. 3 (2007) 69-85. DOI:10.2147/nedt.2007.3.issue-1 |
| [10] |
S.W.B. Yu, S.S.C. Rao, Therap. Adv. Gastroenterol. 7 (2014) 193-205. DOI:10.1177/1756283X14537882 |
| [11] |
R.H. Thomas, D.R. Luthin, Pharmacotherapy 35 (2015) 613-630. DOI:10.1002/phar.2015.35.issue-6 |
| [12] |
K.B. Akondi, M. Muttenthaler, S. Dutertre, et al., Chem. Rev. 114 (2014) 5815-5847. DOI:10.1021/cr400401e |
| [13] |
A.C. Conibear, D.J. Craik, Angew. Chem. Int. Ed. 53 (2014) 10612-10623. DOI:10.1002/anie.201402167 |
| [14] |
B.M. Olivera, W.R. Gray, R. Zeikus, et al., Science 230 (1985) 1338-1343. DOI:10.1126/science.4071055 |
| [15] |
R.A. Myers, L.J.L. Cruz, J.E. Rivier, B.M. Olivera, Chem. Rev. 93 (1993) 1923-1936. DOI:10.1021/cr00021a013 |
| [16] |
A.G. Craig, J. Toxicol. Toxin Rev. 19 (2000) 53-93. DOI:10.1081/TXR-100100315 |
| [17] |
B.M. Ueberheide, D. Fenyö, P.F. Alewood, B.T. Chait, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 6910-6915. DOI:10.1073/pnas.0900745106 |
| [18] |
Q.Y. He, Q.Z. He, X.C. Deng, et al., Nucleic Acids Res. 36 (2008) D293-297. |
| [19] |
G. Naamati, M. Askenazi, M. Linial, Nucleic Acids Res. 37 (2009) W363-368. DOI:10.1093/nar/gkp299 |
| [20] |
E. Lim, A. Pon, Y. Djoumbou, et al., Nucleic Acids Res. 38 (2010) D781-786. DOI:10.1093/nar/gkp934 |
| [21] |
Q. Kaas, R. Yu, A.H. Jin, S. Dutertre, D.J. Craik, Nucleic Acids Res. 40 (2012) D325-330. DOI:10.1093/nar/gkr886 |
| [22] |
J. Gehrmann, N.L. Daly, P.F. Alewood, D.J. Craik, J. Med. Chem. 42 (1999) 2364-2372. DOI:10.1021/jm990114p |
| [23] |
B. Thomma, B. Cammue, K. Thevissen, Planta 216 (2002) 193-202. DOI:10.1007/s00425-002-0902-6 |
| [24] |
T. Ganz, Nat. Rev. Immunol. 3 (2003) 710-720. DOI:10.1038/nri1180 |
| [25] |
R.I. Lehrer, T. Ganz, Curr. Opin. Immunol. 14 (2002) 96-102. DOI:10.1016/S0952-7915(01)00303-X |
| [26] |
D.J. Schibli, H.N. Hunter, V. Aseyev, et al., J. Biol. Chem. 277 (2002) 8279-8289. DOI:10.1074/jbc.M108830200 |
| [27] |
Y.Q. Tang, J. Yuan, G. Ösapay, et al., Science 286 (1999) 498-502. DOI:10.1126/science.286.5439.498 |
| [28] |
M.C. Alexander, T. Hong, L.M. Boo, et al., Proc. Natl. Acad. Sci. U. S. A. 99 (2002) 1813-1818. DOI:10.1073/pnas.052706399 |
| [29] |
M. Trabi, H.J. Schirra, D.J. Craik, Biochemistry 40 (2001) 4211-4221. DOI:10.1021/bi002028t |
| [30] |
A.C. Conibear, K.J. Rosengren, N.L. Daly, S.T. Henriques, D.J. Craik, J. Biol. Chem. 288 (2013) 10830-10840. DOI:10.1074/jbc.M113.451047 |
| [31] |
D.J. Craik, N.L. Daly, T. Bond, C. Waine, J. Mol. Biol. 294 (1999) 1327-1336. DOI:10.1006/jmbi.1999.3383 |
| [32] |
S.T. Henriques, D.J. Craik, ACS Chem. Biol. 7 (2012) 626-636. DOI:10.1021/cb200395f |
| [33] |
C.T.T. Wong, D.K. Rowlands, C.H. Wong, et al., Angew. Chem. Int. Ed. 51 (2012) 5620-5624. DOI:10.1002/anie.201200984 |
| [34] |
K. Jagadish, A. Gould, R. Borra, et al., Angew. Chem. Int. Ed. 54 (2015) 8390-8394. DOI:10.1002/anie.201501186 |
| [35] |
C.K. Wang, C.W. Gruber, M. Cemazar, et al., ACS Chem. Biol. 9 (2014) 156-163. DOI:10.1021/cb400548s |
| [36] |
M.E. Felizmenio-Quimio, N.L. Daly, D.J. Craik, J. Biol. Chem. 276 (2001) 22875-22882. DOI:10.1074/jbc.M101666200 |
| [37] |
C. Jennings, J. West, C. Waine, D. Craik, M. Anderson, Proc. Natl. Acad. Sci. U. S. A. 98 (2001) 10614-10619. DOI:10.1073/pnas.191366898 |
| [38] |
M.L. Colgrave, A.C. Kotze, Y.H. Huang, et al., Biochemistry 47 (2008) 5581-5589. DOI:10.1021/bi800223y |
| [39] |
M.R.R. Plan, I. Saska, A.G. Caguan, D.J. Craik, J. Agric. Food. Chem. 56 (2008) 5237-5241. DOI:10.1021/jf800302f |
| [40] |
B. Chen, M.L. Colgrave, N.L. Daly, et al., J. Biol. Chem. 280 (2005) 22395-22405. DOI:10.1074/jbc.M501737200 |
| [41] |
S.T. Henriques, Y.H. Huang, S. Chaousis, et al., Chem. Biol. 22 (2015) 1087-1097. DOI:10.1016/j.chembiol.2015.07.012 |
| [42] |
H. Terlau, B.M. Olivera, Physiol. Rev. 84 (2004) 41-68. DOI:10.1152/physrev.00020.2003 |
| [43] |
W.R. Gray, B.M. Olivera, G.C. Zafaralla, et al., Biochemistry 31 (1992) 11864-11873. DOI:10.1021/bi00162a027 |
| [44] |
E. Leipold, A. Hansel, B.M. Olivera, H. Terlau, S.H. Heinemann, FEBS Lett. 579 (2005) 3881-3884. DOI:10.1016/j.febslet.2005.05.077 |
| [45] |
K.J. Shon, M. Stocker, H. Terlau, et al., J. Biol. Chem. 273 (1998) 33-38. DOI:10.1074/jbc.273.1.33 |
| [46] |
K.J. Nielsen, T. Schroeder, R. Lewis, J. Mol. Recognit. 13 (2000) 55-70. DOI:10.1002/(ISSN)1099-1352 |
| [47] |
R.A. Li, G.F. Tomaselli, Toxicon 44 (2004) 117-122. DOI:10.1016/j.toxicon.2004.03.028 |
| [48] |
G.P. Miljanich, Curr. Med. Chem. 11 (2004) 3029-3040. DOI:10.2174/0929867043363884 |
| [49] |
L. Moise, A. Piserchio, V.J. Basus, E. Hawrot, J. Biol. Chem. 277 (2002) 12406-12417. DOI:10.1074/jbc.M110320200 |
| [50] |
H.S. Young, L.G. Herbette, V. Skita, Biophys. J. 85 (2003) 943-953. DOI:10.1016/S0006-3495(03)74533-0 |
| [51] |
Y.H. Chen, J.C. Tai, W.J. Huang, et al., Biochemistry 21 (1982) 2592-2600. DOI:10.1021/bi00540a003 |
| [52] |
R. Doley, R.M. Kini, Cell. Mol. Life Sci. 66 (2009) 2851-2871. DOI:10.1007/s00018-009-0050-2 |
| [53] |
Y.C. Cheng, J.J. Wang, L.S. Chang, Toxicon 51 (2008) 304-315. DOI:10.1016/j.toxicon.2007.10.006 |
| [54] |
J.H. Shiu, C.Y. Chen, L.S. Chang, et al., Proteins 57 (2004) 839-849. DOI:10.1002/(ISSN)1097-0134 |
| [55] |
J.C. Dewan, G.A. Grant, J.C. Sacchettini, Biochemistry 33 (1994) 13147-13154. DOI:10.1021/bi00248a026 |
| [56] |
J. Ciolek, H. Reinfrank, L. Quinton, et al., Proc. Natl. Acad. Sci. U. S. A. 114 (2017) 7154-7159. DOI:10.1073/pnas.1620454114 |
| [57] |
S. Diochot, A. Baron, M. Salinas, et al., Nature 490 (2012) 552-555. DOI:10.1038/nature11494 |
| [58] |
J.P. Rosso, J.R. Schwarz, M. Diaz-Bustamante, et al., Proc. Natl. Acad. Sci. U. S. A. 112 (2015) E891-E900. DOI:10.1073/pnas.1415488112 |
| [59] |
P. Hidalgo, R. Mackinnon, Science 268 (1995) 307-310. DOI:10.1126/science.7716527 |
| [60] |
R.C.R. Vega, L.D. Possani, Toxicon 43 (2004) 865-875. DOI:10.1016/j.toxicon.2004.03.022 |
| [61] |
T. Durek, I. Vetter, C.A. Wang, et al., ACS Chem. Biol. 8 (2013) 1215-1222. DOI:10.1021/cb400012k |
| [62] |
J.J. Smith, J.M. Hill, M.J. Little, et al., Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 10478-10483. DOI:10.1073/pnas.1103501108 |
| [63] |
M.L. Ruiz, R.L. Kraus, J. Med. Chem. 58 (2015) 7093-7118. DOI:10.1021/jm501981g |
| [64] |
D.J. Craik, N.L. Daly, C. Waine, Toxicon 39 (2001) 43-60. DOI:10.1016/S0041-0101(00)00160-4 |
| [65] |
J.H. Park, K.P. Carlin, G. Wu, et al., J. Med. Chem. 57 (2014) 6623-6631. DOI:10.1021/jm500687u |
| [66] |
J.D. Osteen, V. Herzig, J. Gilchrist, et al., Nature 534 (2016) 494-499. DOI:10.1038/nature17976 |
| [67] |
J. Siemens, S. Zhou, R. Piskorowski, et al., Nature 444 (2006) 208-212. DOI:10.1038/nature05285 |
| [68] |
C.J. Bohlen, A. Priel, S. Zhou, et al., Cell 141 (2010) 834-845. DOI:10.1016/j.cell.2010.03.052 |
| [69] |
M. Kita, D.S. Black, O. Ohno, et al., J. Am. Chem. Soc. 131 (2009) 18038-18039. DOI:10.1021/ja908148z |
| [70] |
S. Yang, Y. Xiao, D. Kang, et al., Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 17534-17539. DOI:10.1073/pnas.1306285110 |
| [71] |
Z.C. Liu, R. Zhang, F. Zhao, et al., J. Proteome Res. 11 (2012) 6197-6212. DOI:10.1021/pr300881d |
| [72] |
P. un, F. Wu, M. Wen, et al., Sci. Rep. 5 (2015) 13399. DOI:10.1038/srep13399 |
| [73] |
M.J. Gallagher, K.M. Blumenthal, J. Biol. Chem. 269 (1994) 254-259. |
| [74] |
Z. Dekan, S.J. Headey, M. Scanlon, et al., Angew. Chem. Int. Ed. 56 (2017) 8495-8499. DOI:10.1002/anie.201703360 |
| [75] |
E. Habermann, Angew. Chem. Int. Ed. 12 (1973) 83-84. |
| [76] |
A. Gould, Y. Li, S. Majumder, et al., Mol. BioSyst. 8 (2012) 1359-1365. DOI:10.1039/c2mb05451e |
| [77] |
J.S. Zheng, S. Tang, G. Ye, H.N. Chang, L. Liu, ChemBioChem 13 (2012) 542-546. DOI:10.1002/cbic.v13.4 |
| [78] |
J.X. Wang, G.M. Fang, Y. He, et al., Angew. Chem. Int. Ed. 54 (2015) 2194-2198. DOI:10.1002/anie.201408078 |
| [79] |
M. Pan, Y. He, M. Wen, et al., Chem. Commun. 50 (2014) 5837-5839. DOI:10.1039/C4CC00779D |
| [80] |
P.E. Dawson, T.W. Muir, I. Clark-Lewis, S.B.H. Kent, Science 266 (1994) 776-779. DOI:10.1126/science.7973629 |
| [81] |
G.M. Fang, J.X. Wang, L. Liu, Angew. Chem. Int. Ed. 51 (2012) 10347-10350. DOI:10.1002/anie.201203843 |
| [82] |
J.S. Zheng, S. Tang, Y.K. Qi, Z.P. Wang, L. Liu, Nat. Protoc. 8 (2013) 2483-2495. DOI:10.1038/nprot.2013.152 |
| [83] |
J.S. Zheng, H.N. Chang, F.L. Wang, L. Liu, J. Am. Chem. Soc. 133 (2011) 11080-11083. DOI:10.1021/ja204088a |
| [84] |
S. Tang, Y.Y. Si, Z.P. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 5713-5717. DOI:10.1002/anie.201500051 |
| [85] |
G.M. Fang, Y.M. Li, F. Shen, et al., Angew. Chem. Int. Ed. 50 (2011) 7645-7649. DOI:10.1002/anie.201100996 |
| [86] |
Y.C. Huang, Y.M. Li, Y. Chen, et al., Angew. Chem. Int. Ed. 52 (2013) 4858-4862. DOI:10.1002/anie.v52.18 |
| [87] |
Y.M. Li, Y.T. Li, M. Pan, et al., Angew. Chem. Int. Ed. 53 (2014) 2198-2202. DOI:10.1002/anie.201310010 |
| [88] |
Z. Wang, W. Xu, L. Liu, T.F. Zhu, Nat. Chem. 8 (2016) 698-704. DOI:10.1038/nchem.2517 |
| [89] |
M. Pan, S. Gao, Y. Zheng, et al., J. Am. Chem. Soc. 138 (2016) 7429-7435. DOI:10.1021/jacs.6b04031 |
| [90] |
R. Zitterbart, O. Seitz, Angew. Chem. Int. Ed. 55 (2016) 7252-7256. DOI:10.1002/anie.201601843 |
| [91] |
S.F. Loibl, Z. Harpaz, O. Seitz, Angew. Chem. Int. Ed. 54 (2015) 15055-15059. DOI:10.1002/anie.201505274 |
| [92] |
M.M. Altamirano, C. García, L.D. Possani, A.R. Fersht, Nat. Biotechnol. 17 (1999) 187-191. DOI:10.1038/6192 |
| [93] |
M. Sela, F.H. White, C.B. Anfinsen, Science 125 (1957) 691-692. DOI:10.1126/science.125.3250.691 |
| [94] |
G. Bulaj, O. Buczek, I. Goodsell, et al., Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 14562-14568. DOI:10.1073/pnas.2335845100 |
| [95] |
A. Schrimpf, U. Linne, A. Geyer, Org. Biomol. Chem. 15 (2017) 2512-2521. DOI:10.1039/C6OB02746F |
| [96] |
V.P. Terrier, A.F. Delmas, V. Aucagne, Org. Biomol. Chem. 15 (2017) 316-319. DOI:10.1039/C6OB02546C |
| [97] |
C.I. Schroeder, L.D. Rash, X. Vila-Farrés, et al., Angew. Chem. Int. Ed. 53 (2014) 1017-1020. DOI:10.1002/anie.201308898 |
| [98] |
B. Dang, T. Kubota, A.M. Correa, F. Bezanilla, S.B.H. Kent, Angew. Chem. Int. Ed. 53 (2014) 8970-8974. DOI:10.1002/anie.201404438 |
| [99] |
B. Dang, T. Kubota, K. Mandal, et al., Angew. Chem. Int. Ed. 55 (2016) 8639-8642. DOI:10.1002/anie.201603420 |
| [100] |
B. Dang, T. Kubota, K. Mandal, F. Bezanilla, S.B.H. Kent, J. Am. Chem. Soc. 135 (2013) 11911-11919. DOI:10.1021/ja4046795 |
| [101] |
M. Góngora-Benítez, J. Tulla-Puche, F. Albericio, Chem. Rev. 114 (2014) 901-926. DOI:10.1021/cr400031z |
| [102] |
T.M. Postma, F. Albericio, Eur. J. Org. Chem. 17 (2014) 3519-3530. |
| [103] |
H. Hibino, Y. Miki, Y. Nishiuchi, J. Pept. Sci. 20 (2014) 30-35. DOI:10.1002/psc.v20.1 |
| [104] |
Y.K. Qi, S. Tang, Y.C. Huang, et al., Org. Biomol. Chem. 14 (2016) 4194-4198. DOI:10.1039/C6OB00450D |
| [105] |
B.L. Pentelute, S.B.H. Kent, Org. Lett. 9 (2007) 687-690. DOI:10.1021/ol0630144 |
| [106] |
X. Yang, V. Gelfanov, F. Liu, R. DiMarchi, Org. Lett. 18 (2016) 5516-5519. DOI:10.1021/acs.orglett.6b02751 |
| [107] |
S.K. Maity, M. Jbara, S. Laps, A. Brik, Angew. Chem. Int. Ed. 55 (2016) 8108-8112. DOI:10.1002/anie.201603169 |
| [108] |
M. Jbara, S.K. Maity, A. Brik, Angew. Chem. Int. Ed. 56 (2017) 10644-10655. DOI:10.1002/anie.201702370 |
| [109] |
T.D. Kondasinghe, H.Y. Saraha, S.B. Odeesho, J.L. Stockdill, Org. Biomol. Chem. 15 (2017) 2914-2918. DOI:10.1039/C7OB00536A |
| [110] |
T.M. Postma, M. Giraud, F. Albericio, Org. Lett. 14 (2012) 5468-5471. DOI:10.1021/ol3025499 |
| [111] |
M. Muttenthaler, Y.G. Ramos, D. Feytens, A.D. de Araujo, P.F. Alewood, Pept. Sci. 94 (2010) 423-432. DOI:10.1002/bip.21502 |
| [112] |
A. Brust, C.I.A. Wang, N.L. Daly, et al., Angew. Chem. Int. Ed. 52 (2013) 12020-12023. DOI:10.1002/anie.201304660 |
| [113] |
F. Shen, Z.P. Zhang, J.B. Li, Y. Lin, L. Liu, Org. Lett. 13 (2011) 568-571. DOI:10.1021/ol1028755 |
| [114] |
Z. Dekan, M. Mobli, M.W. Pennington, et al., Angew. Chem. Int. Ed. 53 (2014) 2931-2934. DOI:10.1002/anie.v53.11 |
| [115] |
J.L. Stymiest, B.F. Mitchell, S. Wong, J.C. Vederas, Org. Lett. 5 (2003) 47-49. DOI:10.1021/ol027160v |
| [116] |
D.J. Derksen, J.L. Stymiest, J.C. Vederas, J. Am. Chem. Soc. 128 (2006) 14252-14253. DOI:10.1021/ja066203q |
| [117] |
S. Chhabra, A. Belgi, P. Bartels, et al., J. Med. Chem. 57 (2014) 9933-9944. DOI:10.1021/jm501126u |
| [118] |
B.J. van Lierop, S.D. Robinson, S.N. Kompella, et al., ACS Chem. Biol. 8 (2013) 1815-1821. DOI:10.1021/cb4002393 |
| [119] |
A. Glas, D. Bier, G. Hahne, et al., Angew. Chem. Int. Ed. 53 (2014) 2489-2493. DOI:10.1002/anie.201310082 |
| [120] |
A. Glas, T.N. Grossmann, Synletter 26 (2015) 1-5. DOI:10.1055/s-00000083 |
| [121] |
H.K. Cui, Y. Guo, Y. He, et al., Angew. Chem. Int. Ed. 52 (2013) 9558-9562. DOI:10.1002/anie.v52.36 |
| [122] |
Y. Guo, D.M. Sun, F.L. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 14276-14281. DOI:10.1002/anie.201500699 |
| [123] |
Y. Xu, T. Wang, C.J. Guan, et al., Tetrahedron Lett. 58 (2017) 1677-1680. DOI:10.1016/j.tetlet.2017.03.024 |
| [124] |
Y. Guo, C. Liu, H. Song, et al., RSC Adv. 7 (2017) 2110-2114. DOI:10.1039/C6RA26617G |
| [125] |
Y. Wu, Y.H. Li, X. Li, et al., Chem. Sci. 8 (2017) 7368-7373. DOI:10.1039/C7SC02420G |
| [126] |
Y. Xu, L. Xu, Y. Xia, et al., Chem. Commun. 51 (2015) 13189-13192. |
| [127] |
P.J. Knerr, W.A. van der Donk, Ann. Rev. Biochem. 81 (2012) 479-505. DOI:10.1146/annurev-biochem-060110-113521 |
| [128] |
L. Huo, W.A. van der Donk, J. Am. Chem. Soc. 138 (2016) 5254-5257. DOI:10.1021/jacs.6b02513 |
| [129] |
J.P. Mayer, J.R. Heil, J. Zhang, M.C. Munson, Tetrahedron Lett. 36 (1995) 7387-7390. DOI:10.1016/0040-4039(95)01548-5 |
| [130] |
Y. Wang, D.H. Chou, Angew. Chem. Int. Ed. 54 (2015) 10931-10934. DOI:10.1002/anie.201503975 |
| [131] |
K. Hu, H. Geng, Q. Zhang, et al., Angew. Chem. Int. Ed. 55 (2016) 8013-8017. DOI:10.1002/anie.201602806 |
| [132] |
P.J. Knerr, A. Tzekou, D. Ricklin, et al., ACS Chem. Biol. 6 (2011) 753-760. DOI:10.1021/cb2000378 |
| [133] |
B. Zhao, D. Yang, J.H. Wong, et al., ChemBioChem 17 (2016) 1416-1420. DOI:10.1002/cbic.v17.15 |
| [134] |
Z.V.F. Wright, S. McCarthy, R. Dickman, et al., J. Am. Chem. Soc. 139 (2017) 13063-13075. DOI:10.1021/jacs.7b06506 |
| [135] |
C.M.B.K. Kourra, N. Cramer, Chem. Sci. 7 (2016) 7007-7012. DOI:10.1039/C6SC02285E |
| [136] |
H.J. Reich, R.J. Hondal, ACS Chem. Biol. 11 (2016) 821-841. DOI:10.1021/acschembio.6b00031 |
| [137] |
A.D. de Araujo, M. Mobli, J. Castro, et al., Nat. Commun. 5 (2014) 3165. DOI:10.1038/ncomms4165 |
| [138] |
A.D. de Araujo, M. Mobli, G.F. King, P.F. Alewood, Angew. Chem. Int. Ed. 51 (2012) 10298-10302. DOI:10.1002/anie.201204229 |
| [139] |
A.D. de Araujo, B. Callaghan, S.T. Nevin, et al., Angew. Chem. Int. Ed. 50 (2011) 6527-6529. DOI:10.1002/anie.v50.29 |
| [140] |
M. Mobli, A.D. de Araújo, L.K. Lambert, et al., Angew. Chem. Int. Ed. 48 (2009) 9312-9314. DOI:10.1002/anie.v48:49 |
| [141] |
M. Muttenthaler, S.T. Nevin, A.A. Grishin, et al., J. Am. Chem. Soc. 132 (2010) 3514-3522. DOI:10.1021/ja910602h |
| [142] |
N.E. Shepherd, H.N. Hoang, G. Abbenante, D.P. Fairlie, J. Am. Chem. Soc. 127 (2005) 2974-2983. DOI:10.1021/ja0456003 |
| [143] |
H.N. Hoang, R.W. Driver, R.L. Beyer, et al., Angew. Chem. Int. Ed. 55 (2016) 8275-8279. DOI:10.1002/anie.201602079 |
| [144] |
H. Zhao, Q.S. Liu, H. Geng, et al., Angew. Chem. Int. Ed. 55 (2016) 12088-12093. DOI:10.1002/anie.201606833 |
| [145] |
Y. Tian, X. Zeng, J. Li, et al., Chem. Sci. 8 (2017) 7576-7581. DOI:10.1039/C7SC03614K |
| [146] |
H. Zhao, Y. Jiang, Y. Tian, et al., Org. Biomol. Chem. 15 (2017) 459-464. DOI:10.1039/C6OB02501C |
| [147] |
B. Hargittai, N.A. Solé, D.R. Groebe, S.N. Abramson, G. Barany, J. Med. Chem. 43 (2000) 4787-4792. DOI:10.1021/jm990635c |
| [148] |
K.K. Khoo, M.J. Wilson, B.J. Smith, et al., J. Med. Chem. 54 (2011) 7558-7566. DOI:10.1021/jm200839a |
| [149] |
C.W. Tornfe, C. Christensen, M. Meldal, J. Org. Chem. 67 (2002) 3057-3064. DOI:10.1021/jo011148j |
| [150] |
V.V. Rostovtsev, G.L. Green, V.V. Fokin, K.B. Sharpless, Angew. Chem. Int. Ed. 41 (2002) 2596-2599. DOI:10.1002/(ISSN)1521-3773 |
| [151] |
M. Fmpting, O. Avrutina, R. Meusinger, et al., Angew. Chem. Int. Ed. 50 (2011) 5207-5211. DOI:10.1002/anie.v50.22 |
| [152] |
S. Pacifico, A. Kerckhoffs, A.J. Fallow, et al., Org. Biomol. Chem. 15 (2017) 4704-4710. DOI:10.1039/C7OB00959C |
| [153] |
K. Holland-Nell, M. Meldal, Angew. Chem. Int. Ed. 50 (2011) 5204-5206. DOI:10.1002/anie.v50.22 |
| [154] |
A. Gori, C.I.A. Wang, P.J. Harvey, et al., Angew. Chem. Int. Ed. 54 (2015) 1361-1364. DOI:10.1002/anie.201409678 |
| [155] |
M. Empting, O. Avrutina, R. Meusinger, et al., Angew. Chem. Int. Ed. 50 (2011) 5207-5211. DOI:10.1002/anie.v50.22 |
| [156] |
A.M. Spokoyny, Y. Zou, J.J. Ling, et al., J. Am. Chem. Soc. 135 (2013) 5946-5949. DOI:10.1021/ja400119t |
| [157] |
Y. Zheng, L. Zhai, Y. Zhao, C. Wu, J. Am. Chem. Soc. 137 (2015) 15094-15097. DOI:10.1021/jacs.5b10779 |
| [158] |
W. Liu, Y. Zheng, X. Kong, et al., Angew. Chem. Int. Ed. 56 (2017) 4458-4463. DOI:10.1002/anie.201610942 |
| [159] |
Y. Chen, T. Li, J. Li, et al., Org. Biomol. Chem. 15 (2017) 1921-1929. DOI:10.1039/C6OB02786E |
| [160] |
T. Lühmann, S.K. Mong, M.D. Simon, L. Meinel, B.L. Pentelute, Org. Biomol. Chem. 14 (2016) 3345-3349. DOI:10.1039/C6OB00208K |
| [161] |
S. Rapireddy, L. Nhon, R.E. Meehan, et al., J. Am. Chem. Soc. 134 (2012) 4041-4044. DOI:10.1021/ja211867j |
| [162] |
J.A. Getz, O. Cheneval, D.J. Craik, P.S. Daugherty, ACS Chem. Biol. 8 (2013) 1147-1154. DOI:10.1021/cb4000585 |
| [163] |
Y.P. Chang, J. Banerjee, C. Dowell, et al., J. Med. Chem. 57 (2014) 3511-3521. DOI:10.1021/jm500183r |
| [164] |
R. Zhao, H. Dai, N. Mendelman, et al., Proc. Natl. Acad. Sci. U. S. A. 112 (2015) E7013-7021. DOI:10.1073/pnas.1514728112 |
| [165] |
J.S. Zheng, S. Tang, Y.C. Huang, L. Liu, Acc. Chem. Res. 46 (2013) 2475-2484. DOI:10.1021/ar400012w |
| [166] |
Y.C. Huang, G.M. Fang, L. Liu, Natl. Sci. Rev. 3 (2016) 107-116. DOI:10.1093/nsr/nwv072 |
| [167] |
J.B. Li, S. Tang, J.S. Zheng, C.L. Tian, L. Liu, Acc. Chem. Res. 50 (2017) 1143-1153. DOI:10.1021/acs.accounts.7b00001 |
| [168] |
B. Yan, L. Ye, W. Xu, L. Liu, Bioorg. Med. Chem. 25 (2017) 4953-4965. DOI:10.1016/j.bmc.2017.05.020 |
| [169] |
B. Farrow, M. Wong, J. Malette, et al., Angew. Chem. Int. Ed. 54 (2015) 7114-7119. DOI:10.1002/anie.201502451 |
| [170] |
Z. Zhu, A. Shaginian, L.S.C. Grady, et al., ACS Chem. Biol. 13 (2018) 53-59. DOI:10.1021/acschembio.7b00852 |