 2018, Vol. 29
2018, Vol. 29 



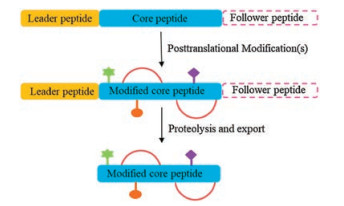
Facilitated by the advance of genome sequencing technology, unprecedented insights have been provided into the genetic capacity of microorganisms to generate bioactive secondary metabolites [1]. Four major classes of natural products have been identified in the past century, including alkaloids, polyketides, terpenoids, and nonribosomal peptides [2]. Peptide natural products are an area of increasing attention for providing leading compounds for current therapeutic challenges, such as "undruggable" protein-protein interactions and antibiotic-resistance of infectious bacteria [3]. Recently, ribosomally synthesized and posttranslationally modified peptides (RiPPs) have emerged as a new family of peptide natural products with diverse bioactivities, such as lantibiotics and thiopeptides [2]. Typically, the biosynthesis of RiPPs requires a precursor peptide, which contains a core peptide fused to a leader peptide or a follower peptide that facilitates the installation of posttranslational modifications (PTMs) (Fig. 1). Upon completion of enzymatic modifications, the leader and/or follower peptide are removed to produce the final product [4]. The extensive modifications of the core peptide provide RiPPs improved resistance to proteolytic degradation and constrained conformation optimal for target binding. A number of RiPPs have advanced to clinical trials, including the thiopeptide LFF571 for the treatment of Clostridium difficile infection [5]. The studies of RiPPs have undergone a revolution in recent years due to the explosion of available genomic data and the genome mining technology that enables the identification of a biosynthetic gene cluster in silico to guide natural product discovery. RiPPs are particularly suited for genome mining because of the direct link between precursor gene and final product and the relatively short biosynthetic pathways, which lend them well for heterologous expression.

|
Download:
|
| Fig. 1. Biosynthesis of RiPPs | |
Thioviridamide-like compounds (Fig. 2) are a subfamily of RiPPs with potent antiproliferative and pro-apoptotic activities. Thioviridamide is first isolated in 2006 from the fermentation of Streptomyces olivoviridis NA005001 [6]. Comprehensive NMR analysis identified the presence of a N-terminal 2-hydroxy-2- methyl-4-oxopentanoyl group, five thioamide groups in the backbone, a β-hydroxy-N1, N3-dimethylhistidinium (hdmHis) residue, and a rare S-[(Z)-2-aminovinyl]-D-cysteine (AviCys) in the C-terminal macrocycle [7]. The biosynthetic machinery and the mode of action of thioviridamide and related compounds remain largely unknown. This review outlined recent advances in the discovery of thioviridamide-like peptide natural products and the elucidation of their biosynthetic origin.

|
Download:
|
| Fig. 2. Structures of thioviridamide-like molecules | |
2. Discovery of thioviridamide-like compounds and their bioactivities
In 2006, Hayakawa et al. reported the isolation and characterization of thioviridamide from S. olivoviridis when screening for antitumor antibiotics using 3Y1 rat fibroblasts transformed with adenovirus oncogenes [6]. In addition to the thioamide groups in the backbone, thioviridamide contains a C-terminal S-(2-aminovinyl) cysteine (AviCys) residue, which is also found in linaridins. For example, epidermin, microbisporicin and cypemycin all contain C-terminal AviCys motifs, which are proposed to derive from the cyclization between a Ser/Cys derived dehydroalanine and a C-terminal Cys via oxidative decarboxylation. Therefore, thioviridamide was proposed to be synthesized following a RiPPs pathway, although the origin of thioamide backbone is not well understood.
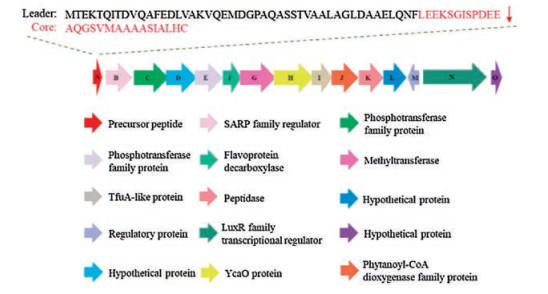
Hayakawa group then sequenced the genomic DNA isolated form S. olivoviridis NA05001. Through gene analysis, they identified a tva gene containing 75 amino acids with a C-terminal sequence of VMAAAASIALHC, which is identical to that of thioviridamide (Fig. 3) [8]. Further analysis of the surrounding genomic region of the tva gene revealed a cluster containing 15 genes arranged as an operon [8]. To identify the essential genes for thioviridamide biosynthesis, Streptomyces lividans TK23 was transformed with the candidate genes for heterologous expression. Indeed, the tva gene cluster (tvaA to tvaO) produced matured thioviridamide in such expression system, conforming its role for thioviridamide production [8]. The antitumor activity of thioviridamide was then evaluated using a variety of cell lines, including 3Y1 rat normal fibroblasts and 3Y1 cells transformed with adenovirus type 12 (Ad12-3Y1), adenovirus E1A gene (E1A-3Y1), SV40 (SV-3Y1), v-src (SR-3Y1) or v-H-ras (HR-3Y1). Results showed that thioviridamide possessed potent cytotoxicity towards Ad12-3Y1 cells (IC50 = 3.9 ng/mL) and E1A-3Y1 cells (IC50 = 32 ng/mL). Recently, a second thioviridamide derivative, JBIR-140, was produced through heterologous expression of a bacterial artificial chromosome (BAC) clone prepared from S. olivoviridis OM13 containing the entire gene cluster for thioviridamide biosynthesis in S. avermitilis SUKA17 strain [9]. Thioviridamide and JBIR-140 were subjected to a number of cytotoxic activity assays, including SKOV-3 (human ovarian adenocarcinoma), Meso-1 (malignant pleural mesothelioma), and Jurkat (immortalized human T lymphocyte). Results showed that both compounds exhibited potent bioactivities against Jurkat, with IC50 values of 12.5 mmol/L and 5.4 mmol/L, respectively (Table 1) [9].

|
Download:
|
| Fig. 3. The tva gene cluster from S. olivoviridis NA05001 | |
|
|
Table 1 Cytotoxic activity of thioviridamide, JBIR-140 and neo-thioviridamide |

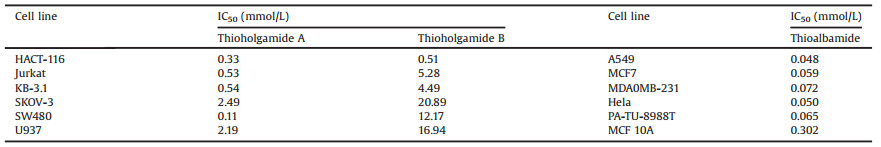
In 2017, Truman and Müller groups independently reported their research on the discovery of thioviridamide-like molecules (TLMs) with the aid of genome mining technology [10]. Truman group used the YcaO-domain protein TvaH as a template for BLAST search in database and analyzed the genomic regions surrounding their respective genes using the tva BGC as the query [10b]. This strategy identified 14 closely related TLM-like BGCs in bacterial genomes. Interestingly, all the gene clusters are identified from actinobacteria, with the exception of the cyanobacterium Mastigocladus laminosus. Five strains were selected, including A. alba DSM 44262, Streptomyces sp. NRRL S-4, Streptomyces sp. NRRL S-15, Streptomyces sp. NRRL S-87, and Nocardiopsis potens DSM 45234, and subject to fermentation in multiple culture conditions. The fermentation extracts were subsequently screened by liquid chromatography-mass spectrometry (LC-MS) for TLM production based on their predicted mass. Four thioviridamide-like molecules (TLMs) were successfully identified using this methodology, including thioholgamide A, thioholgamide B [10a], thioalbamide and thiostreptamide S4 (Fig. 2) [10b]. Thioholgamides A and B were produced by Streptomyces malaysiense MUSC 13657 (DSM 100712), with amino acid substitutions including an N-terminal pyruvyl moiety and four thioamide groups. Surprisingly, the structure of thioholgamide A is highly similar to the natural products thiostreptamide S87 and neo-thioviridamide produced by Streptomyces sp. NRRL S-87 and Streptomyces sp. MSB090213SC12, respectively [11]. Streptomyces sp. NRRL S-4 and Streptomyces sp. NRRL S-15 produced the same compound thiostreptamide S4 with an N-terminal pyruvyl group (Fig. 2). A. alba DSM 44262 produced compound thioalbamide featuring an N-terminal lactyl moiety instead of the 2-hydroxy-2-methyl-4-oxopentanoyl group of thioviridamide. Compared to thioviridamide and JBIR-140, thioholgamide A showed enhanced cytotoxic activity against SKOV-3 and Jurkat with IC50 values of 2.49 mmol/L and 0.53 mmol/L, respectively (Table 2). Thioholgamide B exhibited lower activity compared to thioholgamide A [10a]. Thioalbamides showed strong antiproliferative activity on tumor lines including luminal and basal breast (MDA-MB-231 and MCF7) adenocarcinoma cell lines, pancreatic (PA-TU-8988T), uterine cervical (HeLa) and alveolar (A549) with IC50 values of 0.072, 0.059, 0.065, 0.050, 0.048 mmol/L, respectively [10b].
|
|
Table 2 Cytotoxic activity of thioholgamides and thioalbamide |
{kind=link}
{kind=link}
{kind=link}
3. Biosynthesis of thioviridamide-like compounds
Thioviridamide contains two unique types of posttranslational modifications: thioamides and AviCys. As the biosynthesis of thioviridamide has not been fully elucidated, this review will focus on the origin of these two types of PTMs in nature.
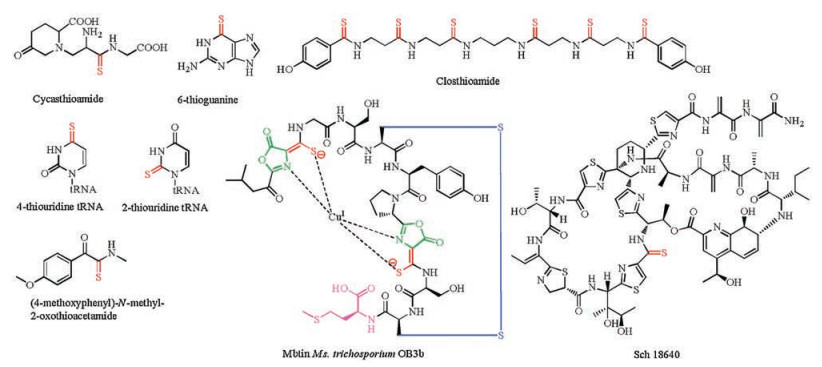
Thioamides are unique alteration of peptide amide bonds by replacing the carbonyl oxygen with sulfur (HN-C=S). The chemical approach for the introduction of thioamide bond into peptides and proteins has been developed to perturb secondary structures, to inhibit proteolysis, as photoswitches, and as spectroscopic labels [12]. Examples of natural products containing thioamides are extremely rare, including thioviridamide-like compounds, methanobactins, closthioamide, Sch18640 and the plant-derived cycasthioamide (Fig. 4) [13]. Thioamide moieties are also found in the structures of nucleotide derivatives such as thiouridine and thioguanine [14]. However, the biosynthesis of thioamides has been obscure.

|
Download:
|
| Fig. 4. Chemical structures of natural products containing thioamides | |
{kind=link}
Recently, an archael ycaO-tfuA gene locus was found to be involved in the post-translational thioamidation at Gly465 in the α-subunit of methyl-coenzyme M reductase (McrA), which is essential in the methanogenic archaeon Methanosarcina acetivorans [15]. Knockout of ycaO, tfuA, and ycaO-tfuA genes of M. acetivorans resulted in the abolishment of thioglycine formation of McrA, indicating the essential role of ycaO-tfuA genes for thioamidation. YcaO enzymes, originally defined as DUF181 (abbreviation of the domains of unknown function), were hypothesized to ubiquitously utilize ATP to O-phosphorylate peptide backbone carbonyl oxygen, generating a common hemiorthoamide intermediate upon nucleophilic attack, eliminating the phosphate to yield formation of azoline, macroamidine, or thioamide during RiPPs biosynthesis [16]. Different types of YcaOs, associated with varied activities, often require different partner proteins, which are typically encoded by the neighboring gene. Among various RiPPs, such as linear azol(in)e-containing peptides, thiopeptides, and cyanobactins, the YcaOs (named azolineforming YcaOs) function together with a member of E1-ubiquitin activating (E1-like) superfamily. Exceptions are the two YcaOs within the antibiotic natural product bottromycin BGC. Bottromycin contains an azoline heterocycle and a macroamidine. Surprisingly, the two YcaOs lack an E1-like partner protein, suggesting that the standalone but fully functional proteins belong to novel categories within the YcaO superfamily (called standalone azoline-forming YcaOs and amidine-forming YcaOs, respectively). In the case of thioviridamide thioamidation, an uncharacterized 'TfuA-like' protein, TvaI, seems to co-occur adjacent to the YcaO gene, TvaH. This distinguishable group is therefore named thioamide-forming YcaOs or TfuA-associated YcaOs. While the role of this fascinating YcaO protein in thioviridamide biosynthesis has yet to be established, based on enzymatic similarity, thioamide installation is believed to share a similar mechanism with azolineforming reaction. As a plausible mechanism, the reaction was initiated by the activation of the carbonyl oxygens by forming Ophosphorylated hemiorthoamide intermediates. Subsequently, a second nucleophile, likely an exogenous source of sulfide (S2-) rather than an adjacent cysteine, serine, or threonine residue, attacks the activated carbonyl by forming a tetrahedral intermediate, which is followed by the elimination of a phosphate group to generate a thioamide motif (Fig. 5) [16]. TfuA partner may contribute to facilitating binding to the peptidic substrate or recruiting and delivering sulfide equivalents directly or indirectly.

|
Download:
|
| Fig. 5. The reactions of thioamides biosynthesis catalyzed by YcaO proteins | |
{kind=link}
In 2018, in vitro investigation by Mitchell and co-workers showed that YcaO and TfuA proteins from M. acetivorans catalyzed the thioglycine formation of McrA sequence (460RLGFFGFDLQD470) in the presence of Na2S and ATP [17]. Two TfuA-independent YcaOs from M. kandleri and M. jannaschii were able to install thioamide on peptidic substrates in the absence of partner proteins. The crystal structures of the M. kandleri YcaO in complex with ADP and ACPMg2+ indicate that the YcaO proteins bind to a single Mg2+ and the nucleotide binds in a similar location. Residues (Glu78, Glu81, Gln160, Glu164, Glu167) involved in Mg2+ coordination and nucleotide-binding in YcaO proteins were further determined by the enzymatic assays in vitro with Ala substituted variants. Thus, these studies have elucidated a new enzymatic transformation by YcaO superfamily, and importantly, lay the foundation for further investigation of the biosynthesis of thioamide-containing compounds.
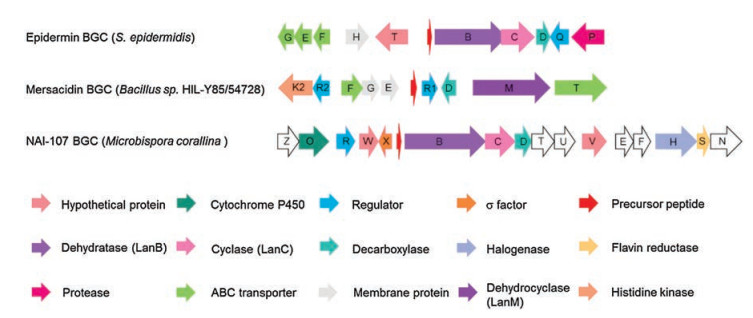
The C-terminal AviCys and AviMeCys residues are the second characteristic posttranslational modification in thioviridamidelike compounds. AviCys residues have been found in three classes of RiPP natural products: lanthipeptides, linaridins, and thioviridamides. Lanthipeptides containing AviCys motifs include epidermin, gallidermin, mutacin 1140, microbisporicin A1 (NAI-107) and mersacidin (Fig. 6) [18]. Linaridins, which do not contain lanthionine bridges but feature an AviCys moiety at their Cterminus, is a small but growing family of RiPPs. Examples from this subfamily include cypemycin and grisemycin (Fig. 7) [19].

|
Download:
|
| Fig. 6. The BGCs of epidermin, mersacidin and NAI-107 | |
{kind=link}

|
Download:
|
| Fig. 7. Structures of natural peptides that contain an AviCys or AviMeCys residue | |
{kind=link}
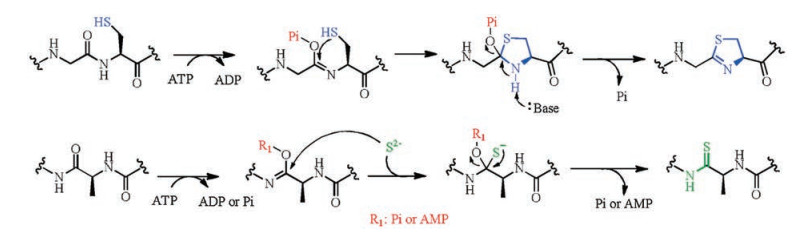
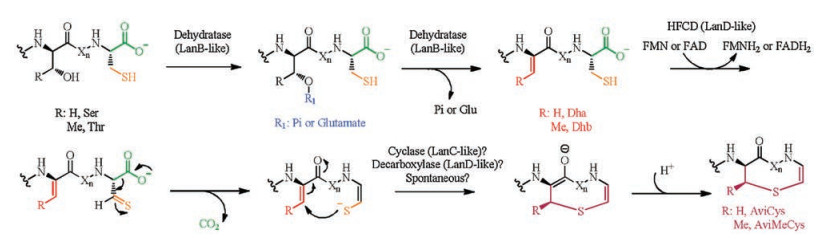
The first example of AviCys containing compounds is epidermin, a lanthipeptide isolated from S. epidermidis [20]. In vitro reconstitution of the putative flavoprotein EpiD from epidermin biosynthetic gene cluster showed its activity to catalyze oxidative decarboxylation of the C-terminal Cys of the precursor peptide EpiA, generating a (Z)-enethiol product [21]. A proposed mechanism delineates the AviCys installation (Fig. 8): the flavin cofactor (FMN or FAD) oxidizes the C-terminal Cys thiol to a thioaldehyde that subsequently delocalizes the negative charge developed at the former Cα upon spontaneous decarboxylation, forming a thioenolate. A nearby Dha or Dhb residue is attacked by the enethioate nucleophile, yielding AviCys or AviMeCys moieties in a similar manner as lanthionine (Lan) or methyl lanthionine (MeLan) formation. However, it remains unclear whether this cyclization reaction is catalyzed by the decarboxylases EpiD, the putative cyclase EpiC, or non-enzymatically. The substrate specificity of EpiD was investigated after the enzymatic activity was determined. A series of experiments showed that it was able to decarboxylate a variety of peptides containing a three amino acid sequence at its C-terminus, where the first amino acid can be V/I/L/ M/F/Y/W and the second amino acid can be A/S/V/T/C/I/L, but the C-terminal residue must be cysteine. In addition to these peptides, some special substrates including the thioether SFNSYCC(Et), the amide SFNSYCC-NH2, and a homocysteine (HCy)-containing heptapeptide, SFNSYCHCy were also examined, but all of these could not be decarboxylated by EpiD. Hence, the free thiol group and carboxyl group of the C-terminal cysteine are essential for the substrate recognition [22].

|
Download:
|
| Fig. 8. A proposed mechanism delineating AviCys installation | |
{kind=link}
Crystal structure of EpiD displays a PASANT motif (residues 81– 86), which is proposed to be the FMN binding motif [20]. Ser83, Asn85 and Thr86 bind to the phosphate group of FMN with their hydrophilic side-chain. Ala84 supports a hydrogen bond to the pyrimidine portion of the isoalloxazine ring. Crystal structure of the inactive mutant H67N of EpiD and pentapeptide DSYTC was also reported. The side chain amide of Asn117 is proximal to one of the β-protons from the C-terminal cysteine of the substrate, supporting the hypothesis that the carbonyl group of Asn117 facilitates the removal of the β-proton of the activated Cys. His67 may serves as an active site base to activate the thiol group and to assist the oxidation of the C-terminal cysteine by generating a thioaldehyde. Even though investigations have been taken to elaborate the mechanism of EpiD, more studies are still needed to explore the exact role of each active site residue.
MrsD from mersacidin biosynthesis binds to a flavin adenine dinucleotide (FAD) and catalyzes the formation of the enethiolate that forms the AviMeCys residue during the biosynthesis of mersacidin [23]. Asn125 and Val76 of MrsD are considered as equivalent to the Asn117 and the hydrophobic residue Ile68 of EpiD during the substrate binding and catalysis, suggesting that the formation of AviCys in epidermin biosynthesis follows the same mechanism of mersacidin biosynthesis. The substrate specificity of MrsD has not been well elucidated yet, however, MrsD selectively decarboxylates MrsA in the presence of EpiA, demonstrating substrate selectivity [22].
MibD from the BGC of compound NAI-107 belongs to the HFCD superfamily and is proposed to function as a decarboxylase [24]. To investigate the substrate specificities of MibD, in vitro assays were carried out with the precursor peptide His6-MibA and the MibA core peptide [24]. Both experiments resulted in a molecular weight loss of 46 Da, which is consistent with the removal of two hydrogen atoms and one molecule of CO2, indicating the leader sequence and the post-translational modifications of the precursor peptide are not required for enzyme recognition. These results raise the possibility that, in all post-translational modifications of NAI-107 biosynthesis, the decarboxylation catalyzed by MibD may act first in vivo. MibD only exhibited activity on its cognate substrate His6- MibA when it was incubated with His6-MibA and other lanthipeptide precursor peptides containing C-terminal FTINVC, LVGKMC, or YWEGEC.
Based on the sequence homology with lanthipeptide Cys oxidative decarboxylase, CypD from the cypemycin cluster of Streptomyces sp. OH-4156 was proposed to be responsible for AviCys formation. Although BGCs of cypemycin do not encode homologs of either lanthipeptide dehydratase (LanB-like) necessary for the installation of the Dha/Dhb or the lanthipeptide cyclase (LanC-like) implicated in AviCys, CypD shows sequence homology with (R)-4'-phospho-N-pantothenoyl-cysteine (PPC) decarboxylases, which is involved in the biosynthesis of 4'-phosphopantetheine in coenzyme A biosynthesis. This class of proteins has been named homo-oligomeric flavin-containing Cys decarboxylase (HFCD) family that employs a flavin cofactor to catalyze the oxidative decarboxylation of C-terminal Cys-containing peptides. The ∆cypD knockout strain produced a nondecarboxylated cypemycin analog retaining all the other PTMs, indicating that CypD is solely involved in decarboxylation of the C-terminal Cys residue. Such a cypemycin analog loses the inhibitory activity towards M. luteus, indicating the importance of AviCys for its antimicrobial activity [25].
4. Genome mining of the gene cluster of thioviridamide-like compoundsAlthough current investigation of the biosynthesis of thioviridamide-like compounds have not fully elucidated the biological logic of their production, however, two key types of enzymes, YcaOs and Flavin-dependent decarboxylases, appear to be essential and characteristic in their biosynthetic gene clusters. Recently, a BLAST search of the YcaO domain protein TvaH identified 22 homologous proteins. Further analysis of their surrounding genes leads to the discovery of 14 closely related BGCs of thioviridamide-like compounds [10b]. Similarly, an effort to identify homologous BGCs from the public database of microorganisms through genome mining lead to the discovery of 13 BGCs which closely resemble the thioviridamide BGC (Fig. 9) [10a]. These BGCs show part of conserved tailoring genes and encode putative precursor peptides which are of high sequence similarity to TvaA, the precursor peptide of thioviridamide. Homologs of eight thioviridamide genes, including tvaA, tvaC, tvaD, tvaE, tvaF, tvaG, tvaH and tvaI, are found in all 13 BGCs. Homologs of tvaJ, tvaK and tvaL genes are found in 12 of 13 genomes. Other genes from thioviridamide biosynthesis, such as tvaB, tvaM, tvaN and tvaO are missing in these genomes. As the gene products of S. olivoridis NA05001, TvaF is proposed to be a flavin-dependent decarboxylase which shows homology to MibD (39% identity, 57% similarity), CypD (27% identity, 49% similarity) and EpiD (25% identity, 49% similarity) [8]. TvaF is proposed to catalyze the post-translational modification of the precursor peptide to generate an AviCys at the C-terminal end of the mature natural product. Two of the proteins encoded by this gene cluster, TvaG and TvaJ, are probably demanded for the structure of the β-hydroxy-N1, N3-dimethylhistidinium (hdmHis) residue. TvaH is classified as the member of YcaO superfamily and thought to catalyze the thioamide synthesis. TvaI, the protein encoded next to TvaH, is annotated as a "TfuA-like" protein and may recruit a sulfurtransferase protein to enhance the thioamidation reaction [16]. Finally, guided by these genome mining efforts, four novel compounds, thioholgamide A, thioholgamide B, thioalbamide and thiostreptamide S4, were isolated and characterized. These studies expand the scope of thioviridamide-like compounds and provide a highly efficient strategy for the discovery of this type of molecules.

|
Download:
|
| Fig. 9. Thioviridamide biosynthetic gene cluster Streptomyces olivoridis NA05001 compared to homologous gene clusters. A, precursor peptide; B, regulatory protein; C, phosphotransferase; D, hypothetical protein; E, phosphotransferase; F, putative flavoprotein decarboxylase; G, putative methyltransferase; H, Ycao-like protein; I, TfuA-like core domain-containing protein; J, putative phytanoyl-CoA dioxygenase family protein; K, putative proteinase; L, membrane protein; M, putative regulatory protein; N, putative LuxR family transcriptional regulator; O, hypothetical protein; X, putative SDR family oxidoreductase; Y, putative methyltransferase; IS, putative IS5/IS1182 family transposase; GAP, sequencing gap | |
{kind=link}
5. Summary
In summary, thioviridamide-like molecules (TLMs) are RiPPs that bear rare posttranslational modifications of thioamides and AviCys. Members of this family usually possess anti-proliferative and pro-apoptotic activities. Two key enzymes, YcaO and CypDlike decarboxylases, are identified from the BGCs of TLMs and are responsible for the formation of thioamides and AviCys motifs. Genome mining of gene clusters of TLMs leads to the discovery of a number of novel thioviridamide-like molecules. The fascinating questions on the mechanism of thioamides and AviCys biosynthesis remain unclear and will be the focus of studies in the future.
AcknowledgmentsThis work is supported by the 1000-Youth Talents Plan, NSF of China (No. 21778030), Natural Science Foundation of Jiangsu Province (No. BK20160640) and the Fundamental Research Funds for the Central Universities.
| [1] |
T.J. Oman, W.A. van der Donk, Nat. Chem. Biol. 6 (2010) 9-18. DOI:10.1038/nchembio.286 |
| [2] |
P.G. Arnison, M.J. Bibb, G. Bierbaum, et al., Nat. Prod. Rep. 30 (2013) 108-160. DOI:10.1039/C2NP20085F |
| [3] |
(a) K. A. Brogden, Nat. Rev. Microbiol. 3 (2005) 238-250.
|
| [4] |
M.A. Ortega, W.A. van der Donk, Cell Chem. Biol. 23 (2016) 31-44. DOI:10.1016/j.chembiol.2015.11.012 |
| [5] |
K. Mullane, C. Lee, A. Bressler, et al., Antimicrob. Agents Chemother. 59 (2015) 1435-1440. DOI:10.1128/AAC.04251-14 |
| [6] |
Y. Hayakawa, K. Sasaki, H. Adachi, et al., J. Antibiot. 59 (2006) 1-5. DOI:10.1038/ja.2006.1 |
| [7] |
Y. Hayakawa, K. Sasaki, K. Nagai, K. Shin-ya, K. Furihata, J. Antibiot. 59 (2006) 6-10. DOI:10.1038/ja.2006.2 |
| [8] |
M. Izawa, T. Kawasaki, Y. Hayakawa, Appl. Environ. Microbiol. 79 (2013) 7110-7113. DOI:10.1128/AEM.01978-13 |
| [9] |
M. Izumikawa, I. Kozone, J. Hashimoto, et al., J. Antibiot. 68 (2015) 533-536. DOI:10.1038/ja.2015.20 |
| [10] |
(a) L. Kjaerulff, A. Sikandar, N. Zaburannyi, et al., ACS Chem. Biol. 12 (2017) 2837-2841.
|
| [11] |
T. Kawahara, M. Izumikawa, I. Kozone, et al., J. Nat. Prod. 81 (2018) 264-269. DOI:10.1021/acs.jnatprod.7b00607 |
| [12] |
(a) S. Batjargal, Y. J. Wang, J. M. Goldberg, et al., J. Am. Chem. Soc. 134 (2012) 9172-9182.
|
| [13] |
S. Banala, R.D. Sussmuth, Chembiochem 11 (2010) 1335-1337. DOI:10.1002/cbic.201000266 |
| [14] |
K.L. Dunbar, D.H. Scharf, A. Litomska, C. Hertweck, Chem. Rev. 117 (2017) 5521-5577. DOI:10.1021/acs.chemrev.6b00697 |
| [15] |
D.D. Nayak, N. Mahanta, D.A. Mitchell, W.W. Metcalf, Elife pii (2017) e29218. |
| [16] |
B.J. Burkhart, C.J. Schwalen, G. Mann, J.H. Naismith, D.A. Mitchell, Chem. Rev. 117 (2017) 5389-5456. DOI:10.1021/acs.chemrev.6b00623 |
| [17] |
N. Mahanta, A. Liu, S. Dong, S.K. Nair, D.A. Mitchell, Proc. Natl. Acad. Sci. (2018). DOI:10.1073/pnas.1722324115 |
| [18] |
L.M. Repka, J.R. Chekan, S.K. Nair, W.A. van der Donk, Chem. Rev. 117 (2017) 5457-5520. DOI:10.1021/acs.chemrev.6b00591 |
| [19] |
T. Mo, W.Q. Liu, W. Ji, et al., ACS Chem. Biol. 12 (2017) 1484-1488. DOI:10.1021/acschembio.7b00262 |
| [20] |
M. Blaesse, T. Kupke, R. Huber, S. Steinbacher, Embo J. 19 (2000) 6299-6310. DOI:10.1093/emboj/19.23.6299 |
| [21] |
T. Kupke, C. Kempter, V. Gnau, G. Jung, F. Gotz, J. Biol. Chem. 269 (1994) 5653-5659. |
| [22] |
C.S. Sit, S. Yoganathan, J.C. Vederas, Acc. Chem. Res. 44 (2011) 261-268. DOI:10.1021/ar1001395 |
| [23] |
M. Blaesse, T. Kupke, R. Huber, S. Steinbacher, Acta Crystallogr. D. Biol. Crystallogr. 59 (2003) 1414-1421. DOI:10.1107/S0907444903011831 |
| [24] |
M.A. Ortega, D.P. Cogan, S. Mukherjee, et al., ACS Chem. Biol. 12 (2017) 548-557. DOI:10.1021/acschembio.6b01031 |
| [25] |
J. Claesen, M. Bibb, Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 16297-16302. DOI:10.1073/pnas.1008608107 |