 2018, Vol. 29
2018, Vol. 29 



Proteins are among the most diverse and versatile class of molecules in nature. Building up on a defined group of monomers, i.e., the 20 natural amino acids, proteins exhibit a vast array of functions in living systems, including mediating metabolic reaction [1], signal transduction [2, 3], molecule transportation [4-6], or simply as structural component [7]. Efficient strategies that add probes—site-selectively—to the proteins are indispensible to the study and control of their functions in complex settings [8-11]. Further, the increasing demands on proteins or peptides as drugs or functional materials have greatly speeded up the methodology development for protein/peptide ligation or modification [12-14]. Broadly speaking, there are two ways of manipulating proteins, i.e., chemically or enzymatically. The chemical approaches usually harness the reactivity of protein side-chains of certain amino acid residues, e.g., Cys, Lys or Tyr, and have contributed significantly in protein chemical biology [8, 9, 15, 16]. They are, however, in many cases either not highly site-selective or require an excess addition of the probe molecules [11, 17]. Enzyme-mediated protein modifications, normally based on recognition sequence motifs, are highly site-selective and have been increasingly applied [18-21]. The most important recent discoveries and advancements in applying enzymatic strategies for protein modification are covered in the current review, including sortases, butelase 1, subtilisin related ligases. Selected examples of prominent enzymes used for protein bioconjugation are also discussed. This is by no means an exhaustive list [22-25]; rather, the current review aims to serve practitioners in designing the most appropriate enzymatic approaches based on their research purposes and laboratory settings.
2. SortasesMany Gram-positive bacteria display proteins on their cell wall that may be vital for colonization and pathogenesis [26]. Sortrases catalyze a transpeptidation reaction anchoring surface proteins, via a sorting signal, to the peptidoglycans of the cell wall [27-29]. As such, sortases have been increasingly important drug targets as they play central role in virulence [30, 31]; they have also emerged as a versatile tool for chemical biologists in manipulating proteins and peptides [32-35]. To this end, the most widely used enzyme of the family is the sortase A from Staphylococcus aureus (SrtAstaph), which recognizes a reaction partner bearing a C-terminal motif LPXTG (X = any amino acid) and cleaves between the Thr and Gly to generate a thioester-linked acyl enzyme intermediate (Fig. 1). The intermediate is then reacted with the other reaction partner bearing an N-terminal Gly, forming a native amide bond. SrtA is a soluble and robust enzyme, and it can be easily obtained either commercially or from recombinant expression with high yields (~40 mg/L) [35, 36]. The substrates, be it peptides or proteins, are also easily accessible either via synthetic or recombinant strategies, as such, the SrtAstaph mediated ligation has been very attractive for routine laboratory uses [20]. SrtAstaph has been exploited for a wide range of applications in protein/peptide ligation [37, 38], protein/peptide labeling [39], living-cell surface modification [40], and protein/peptide cyclization [41, 42]. There have been many excellent reviews in the literature highlighting the application of sortases, which provide a more comprehensive discussion [35, 43, 44].

|
Download:
|
| Fig. 1. Schematic representation of the sortases mediated protein/peptide modification. Note that when (G)n is placed at the N-terminal of substrate 1, the reaction affords a cyclized product | |
{kind=link}
Initially, the wild-type (wt) SrtAstaph is the go-to sortase enzyme, in which the extremely soluble variants lacking the first 25 or 59 residues are among the most frequent choices. These variants are easy to obtain recombinantly and their structural and biochemical features have been well characterized. It establishes His120–Cys184–Arg197 as the catalytic triad, while Cys184 plays a key role by forming a thioester bond between the enzyme and the LPXTG motif (Fig. 1). The catalytic efficiency (kcat/KM = 50 L mol-1 s-1) of SrtAstaph, however, is generally low, partly because of the high KM value (up to the mmol/L range) and the instability of the thioester intermediate (e.g., hydrolysis, Fig. 1) [35, 45]. Typically, a high enzyme/substrate ratio (up to 1:1) or an extended reaction time (up to one day) would be necessary to obtain satisfactory results [36, 46]. Moreover, the use of SrtAstaph for peptide/protein modification has draw skepticism by other factors such as the reaction reversibility (i.e., breaking of the newly-formed amide bond) and the requirement of Ca2+ in the reaction (mmol/L range), which renders it's in vivo application problematic.
Significant progresses have been made to circumvent the limitations of SrtAstaph. A notable example is the recent identification of a so-called SrtAstaph pentamutant where five mutations (P94R/D160N/D165A/K190E/K196T) have been introduced through directed evolution. The pentamutant shows a ~120-fold increased enzymatic activity compared to the wt SrtAstaph [47]. Combing the previously reported dual mutants, E105K/E108A or E105K/E108Q, that could lift the Ca2+-dependence [48], SrtAstaph heptamutants have been developed to achieve high enzymatic efficiency in the absence of Ca2+ [49, 50]. At present, these heptmutants arguably represent the most universally applicable sortase variants that have already seen increasing use in demanding processes such as live-cell labeling and intracellular ligations [40, 51, 52].
As far as the reversibility of the sortase reaction is concerned, the most obvious (maybe not so economic) solution would be the addition of a huge excess of one of the reaction partners, should it be easily accessible or inexpensive. A complementary approach is the removing of reaction by-product, i.e., the aminoglycine peptide fragment [53]. There are also a number of studies on the design of the reaction partners that render the sortase ligation irreversible. A notable example is the use of modified depsipeptide substrates that upon ligation release nonreactive fragments, e.g., a nonreactive hydroxyacetate moiety [54, 55], or spontaneously form a diketopiperazine [56]. Additionally, the use of substrates such as hydrazine and its derivatives could lead to irreversible protein hydrazinolysis, and hence protein ligation [57]. Finally, there are a number of options available to bypass the inherent need of the LPXTG sequence as the sortase recognition motif. One of the approaches is the use of natural sortase homologs of different bacterial origins. For example, sortase A from Streptococcus pyogenes is Ca2+-independent and known to accept both LPXTG and LPXTA [58, 59].
As a final remark, in setting up a sortase ligation, it is advisable to consider, at least, the following factors: 1) the influence of the recognition motifs, e.g., LPXTG, on the modified proteins or peptides; 2) the accessibility of the reactive termini (both the Cterminal LPXTG motif and N-terminal glycine); 3) ideally, the protein targets to be labeled should be the limiting agent, this way the other reaction partner—usually smaller in size and easier to obtain—can be used in excess to ensure a full conversion; 4) choose the right sortase variants depending on whether the presence of Ca2+ is a problem for the reaction.
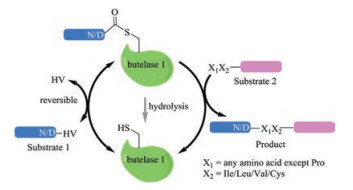
3. Butelase 1While the use of sortases raises concerns over the need of an extra LPTXG motif and low catalytic efficiency, a promising alternative ligase—butelase 1, has been recently reported. Butelase 1 is an Asn/Asp (Asx)-specific cysteine transpeptidase derived from the butterfly pea (Clitoria ternatea), which facilitates the biosynthesis of cyclotides in nature [60]. It only requires a simple Cterminal recognition motif of N/D-HV, and the HV dipeptide will be cleaved, leading to an Asx–butelase 1 thioester intermediate complex (Fig. 2). As for the incoming ligation partner, almost any proteinogenic amino acid at its N-termini may be tolerated by butelase 1, except Pro; moreover, the second amino acid needs to be hydrophobic (e.g., Cys, Ile, Leu or Val). No part of the recognition motif, except for Asx, is left behind in the cyclized or ligated products, which renders the reaction almost "traceless" (Fig. 2). Most strikingly, butelase 1 shows an unpaired catalytic efficiency, with a kcat/KM reaching as high as 1340000 L mol-1 s-1 for medium-sized peptides and incredible cyclization rates that are >10000 times faster than those of other ligases, e.g., sortases [61]. As such, ever since its first publication in 2014, butelase 1 has quickly evolved as a powerful tool in protein modification and engineering [62], with applications already documented in peptide/protein ligation and labeling [60, 63], peptide/protein macrocyclization [61, 64], and living-cell surface labeling [65].

|
Download:
|
| Fig. 2. Schematic representation of the butelase 1 mediated protein/peptide modification. Note that when X1X2 is placed at the N-terminal of substrate 1, the reaction affords a cyclized product | |
{kind=link}
The butelase 1 mediate ligation is convenient to execute in standard biochemical laboratory settings. Typically, the substrate can be readily prepared via chemical synthesis or recombinant expression, and the enzyme is usuallyadded invery small quantities (approx. 0.005 molar equiv.) [62]. There are, however, a number of limitations that need to be addressed for an even broader application of butelase 1. A major obstacle is the availability of the enzyme, as currently, butelase 1 can only be obtained from C. ternatea via alaborious extraction and theyield is ~ 5 mg/kgoffresh plant. While the recombinant expression of butelase 1 has not been successful, a less active homolog of butelase 1, OaAEP1b, was recently recombinnatly expressed, albeit a lowyield (< 2 mg/L) [66]. The X-ray crystal structure of another enzyme of the same family, OaAEP1, has been solved, which may serve as a template to understand the catalyticmechanism of the Asx ligases and facilitate the design of more effective enzymes with even broader substrate scope [67]. Other issues need to be considered when choosing the butelase 1 mediated ligation include: 1) for intermolecular ligation, one of the reaction partners has to be used in excess (>5 equiv.), otherwise, the ligation is reversible (Fig. 2). The use of thiodepsipeptide as a substrate proved to be a potential solution, but the other substrate still has to be added in a small excess [68]; 2) Althoughhasbeen shown to cyclize proteinsbeyond200 amino acid residues very effectively, butelase 1 is not efficient when used for the cyclization of small peptides (e.g., < 9 amino acid residues) and denatured proteins/peptides [62].
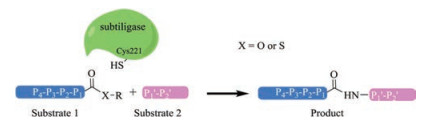
4. Subtilisin related ligasesSubtilisin is a family of nonspecific serine proteases that usually cleaves the protein/peptide with a serine residue at the active site [69]. They are often obtained from soil bacterial, e.g., in large quantities from Bacillus amyloliquefaciens [70]. Intrigued by the long-standing interests of using serine proteases in peptide synthesis, a double mutant of subtilisin BPN' B. amyloliquefaciens have been designed where the catalytic Ser residue was mutated to Cys and a bulky Pro residue at the catalytic center was mutated to Ala [71]. The double mutated subtilisin BPN', termed subtiligase, has an alter mechanism in which the aminolysis is favored over peptidase activity, thereby leading to peptide ligation [71]. Subtiligase has been successfully applied in a number of areas including peptide/protein ligation [71-74] and cyclization [75].
Subtiligase mediates the ligation between a peptide C-terminal ester and a peptide N-terminal α-amine, unlike sortases and butelase 1, it does not require a certain recognition motif at the termini of any reaction partners. However, the sequence properties of the substrates do have great influence on the catalytic performance (kcat/KM) of subtiligase, as an efficient binding between the pair is essential for the reaction. It has been shown that the P4, P1, P10 and P20 positions are critical for ligation efficiency (Fig. 3). Typically, the sequence of each target protein/ peptide and modifier pair has to be optimized to obtain satisfactory yields, and this could be a tedious work. Meanwhile, the need of a large excess of one ligation partner and the presence of Ca2+ in the ligation pose further challenges for the use of subtiligase. Nevertheless, great progresses have been made towards broadening of their application, where both the engineering of substrates [72] and the enzyme itself [76, 77] are reported. To this end, a noticeable development is the recent report by Cole et al. showing a subtiligase variant (Y217 K) capable of catalyzing the expressed protein ligation between a protein thioester and a Cys-free peptide, which renders subtiligase a versatile tool in protein semisynthesis [78]. Finally, a truncated subtilisin BPN' variant, peptiligase, which is Ca2+-independent and stable in the presence of organic solvent and denaturing agents, has been shown to afford extremely high ligation yield (>98%) with only a slight excess of one of the reaction partners [79]. Peptiligase variants (e.g., the commercially available omniligase-1) have been applied in gram-scale peptide synthesis with high yields, making them viable industrial enzymes [80-82].

|
Download:
|
| Fig. 3. Schematic representation of the interaction of the active site of the subtilisin-derived subtiligase with the N-terminal P1-P4 and C-terminal P10-P20 of the substrates | |
{kind=link}
5. Enzymes used in protein/peptide bioconjugation
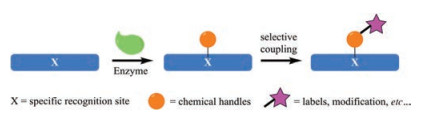
Beside the aforementioned sortases, butelase 1 and subtiligase, many other enzymes have been exploited especially for sitespecific incorporation of bio-orthogonal functionalities (or handles) on proteins/peptides, but mainly via a two-step process [83]. They firstly recognize a defined peptide sequence (tag) and introduce chemical modifications on a specific amino acid residue (e.g., Cys, Lys, Gln or Ser) within the sequence. These chemical modifications could then be further elaborated through highly efficient coupling reactions, affording the desired outcomes, e.g., site-specific protein labeling (Fig. 4).

|
Download:
|
| Fig. 4. Schematic representation of a two-step process of enzyme-mediated protein labeling | |
{kind=link}
5.1. Formylglycine generating enzyme
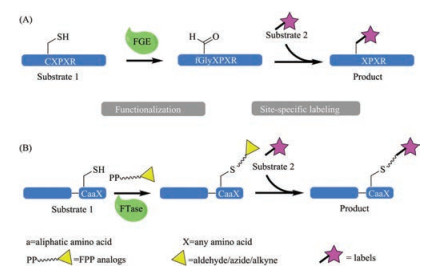
One example is the formylglycine generating enzyme (FGE) that recognizes a hexapeptide sequence LCTPSR and converts the cysteine residue to a aldehyde bearing formylglycine [84-86], a key modification for the activity of the human type I sulfatases [87]. Bertozzi and co-workers have since identified a general consensus sequence motif of CXPXR (Fig. 5A), termed aldehyde tag, and harnessed the versatile reactivity of the resulting aldehyde group for protein labeling in a number of experimental settings, both in vitro and in vivo [88, 89]. It is worth noting that FGEs of some bacterial origins (e.g., Mycobacterium tuberculosis or E. coli) could recognize non canonical sequence motif, which expands the range of aldehyde tag sequences for protein engineering [90]. The great advantage of the FGE strategy is its flexibility: the aldehyde tag can be attached to the N- or C- terminal of the protein of interest, as well as internally, provided that the relevant sites are solvent accessible; it is applicable in both eukaryotic and prokaryotic cells, both in vitro and in vivo; the number of aldehyde group chemistries, e.g., oximation and hydrazine Pictet–Spengler [91, 92]. As such, FGE is a very promising enzyme for protein bioconjugation, and has already been used in a variety of applications, e.g., site-specific protein PEGylation [88], protein immobilization [93], protein glycosylation [94], protin–DNA conjugates and antibody-drug conjugates [95, 96]. It should be noted, however, the influence of the aldehyde-tag (6-13 residues in length) on the properties of the target proteins and the CuII that required for the activity of reconstituted FGE [97] must be evaluated. Moreover, the need of an excess of aldehyde reactive compounds (up to 20 equiv.) and a prolonged reaction time (up to 16 h) would be expected for the second labeling step [91].

|
Download:
|
| Fig. 5. Schematic representation of a two-step process of protein labeling mediated by FGE (A) and FTase (B) | |
{kind=link}
5.2. Farnesyltransferase
Farnesyltransferase (FTase) belongs to a family of three prenyltransferase enzymes [98]. FTase transfers the farnesyl group of farnesyl pyrophosphate (FPP) to a substrate protein, i.e., to the Cys of the C-terminal tetrapeptide sequence motif, CaaX (C is cysteine, a is an aliphatic amino acid and X can be a variety of amino acids) (Fig. 5B) [98, 99]. A well-known substrate is Ras, whose farnesylation is a prerequisite for its cell-transforming potential, and FTase inhibitors have since been hot targets in developing anticancer agents [100-103]. Nevertheless, recent years have seen a growing number of reports in using FTase for protein bioconjugation, because of its flexibility. FTase can tolerate a wide range of FPP analogues carrying a number of functional groups [99], including azide [104], alkyne [105], aldehyde [106] or aryl groups [107]. The corresponding target proteins, now armed with reactive groups, have subsequently been labeled, siteselectively, with certain probes for a number of purposes such as protein immobilization [108, 109] or purification [110]. Moreover, the CaaX motif can be conveniently introduced into the Cterminal of a protein genetically, making virtually any protein a potential substrate for FTase. It should be noted, however, that the influence of the introduction of the CaaX motif and the rather long farnesyl group on the target protein should be closely monitored (such as solubility or activity). Similar to FGE, the FTase mediated bioconjugation is a two-step process, which inherently slows down the overall labeling process and lowers the yield. To this end, efforts have been made to generate more competent FTase variants through protein engineering [111]. Lastly, and very obviously, the FTase strategy can only be used for C-terminal protein modification.
There are other enzymes that have also been exploited for protein bioconjugation, the important ones including lipoic acid ligase [112, 113], biotin ligase [114, 115], tubulin tyrosine ligase [116], transglutaminase [117, 118] and N-myristoyltransferase [119], and we refer the readers to many excellent reviews cited herein.
6. ConclusionIn an ideal situation, one should be able to manipulate proteins/ peptides in a highly controlled (site-/chemo-selective) manner within a complex environment, e.g., living cells. The process should be quick and robust enough to allow the investigation of even the most transient biological pathways. The enzyme-mediated processes show great advantages in selectivity and catalytic efficiency, and a large enzyme toolbox is currently available at the hands of practitioners. As discussed above each enzyme, however, have its own unique properties (such as substrate specificity, size of tag and modification site) or shortcomings. There is no simple solution for all. Therefore, it is advisable to consider the pros and cons of each enzyme before making a decision of the enzyme to be used. Depending on the purpose of the proteinmanipulation, whether it's a proteinlabeling (N-/C- terminal orinternally) or a protein ligation/ cyclization, whether it's for in vitro or in vivo application, different types of enzymes would certainly be required. Secondly, the kinetic parameters of the enzymes, for example, if a fast enzyme kinetic is required, butelase 1 (kcat/KM = 1, 340, 000 L mol-1 s-1) would be a better choice than sortases (kcat/KM = 50 L mol-1 s-1). The accessibility of the enzyme of interest is also an issue, and those that are commercially or recombinantly available should always be the first options. The influence of the length of recognition motifs on the structure and properties of the proteins of interests should be fully accessed, e.g., the C-terminal 4-mer tag CaaX of FTase is generally notexpected toperturb the targetprotein. Similarly, the size and the synthetic difficulties of the probes are also important factors to consider. In many cases, it is not straightforward to predict the outcome of a specific enzyme-mediated protein modification, and several approaches may have to be explored to decide the optimum conditions in applying the transformations [24].
AcknowledgmentsThe financial support from the National Recruitment Program of Global Youth Experts (1000 Talents Plan) and the National Natural Science Foundation of China (No. 81703406) are greatly acknowledged.
| [1] |
A. Bairoch, Nucl. Acids Res. 28 (2000) 304-305. DOI:10.1093/nar/28.1.304 |
| [2] |
L.C. Cantley, K.R. Auger, C. Carpenter, et al., Cell 64 (1991) 281-302. DOI:10.1016/0092-8674(91)90639-G |
| [3] |
S. Mouillet-Richard, M. Ermonval, C. Chebassier, et al., Science 289 (2000) 1925-1928. DOI:10.1126/science.289.5486.1925 |
| [4] |
C. He, M. Knipp, J. Am. Chem. Soc. 131 (2009) 12042-12043. DOI:10.1021/ja9040362 |
| [5] |
F.A. Walker, J. Inorg. Biochem. 99 (2005) 216-236. DOI:10.1016/j.jinorgbio.2004.10.009 |
| [6] |
R.H. Austin, K.W. Beeson, L. Eisenstein, H. Frauenfelder, I.C. Gunsalus, Biochemistry 14 (1975) 5355-5373. DOI:10.1021/bi00695a021 |
| [7] |
P. Fratzl, Collagen: Structure and mechanics, an introduction, in: P. Fratzl (Ed.), Collagen: Structure and Mechanics, Springer US, Boston, MA, 2008, pp. 1-13.
|
| [8] |
C.D. Spicer, B.G. Davis, Nat. Commun. 5 (2014) 4740. DOI:10.1038/ncomms5740 |
| [9] |
O. Boutureira, G.J.L. Bernardes, Chem. Rev. 115 (2015) 2174-2195. DOI:10.1021/cr500399p |
| [10] |
T.L. Foley, M.D. Burkart, Curr. Opin. Chem. Biol. 11 (2007) 12-19. DOI:10.1016/j.cbpa.2006.11.036 |
| [11] |
D. Rabuka, Curr. Opin. Chem. Biol. 14 (2010) 790-796. DOI:10.1016/j.cbpa.2010.09.020 |
| [12] |
J.R. Junutula, H. Raab, S. Clark, et al., Nat. Biotechnol. 26 (2008) 925-932. DOI:10.1038/nbt.1480 |
| [13] |
F.M. Veronese, Biomaterials 22 (2001) 405-417. DOI:10.1016/S0142-9612(00)00193-9 |
| [14] |
G.G. Kochendoerfer, Curr. Opin. Chem. Biol. 9 (2005) 555-560. DOI:10.1016/j.cbpa.2005.10.007 |
| [15] |
J.N. deGruyter, L.R. Malins, P.S. Baran, Biochemistry 56 (2017) 3863-3873. DOI:10.1021/acs.biochem.7b00536 |
| [16] |
M.J. Matos, B.L. Oliveira, N. Martínez-Sáez, et al., J. Am. Chem. Soc. 140 (2018) 4004-4017. DOI:10.1021/jacs.7b12874 |
| [17] |
N. Krall, F.P. da Cruz, O. Boutureira, G.J.L. Bernardes, Nat. Chem. 8 (2015) 103-113. |
| [18] |
E.M. Milczek, Chem. Rev. 118 (2018) 119-141. DOI:10.1021/acs.chemrev.6b00832 |
| [19] |
M. Schmidt, A. Toplak, P.J.L.M. Quaedflieg, J.H. van Maarseveen, T. Nuijens, Drug Discovery Today:Technol. 26 (2017) 11-16. DOI:10.1016/j.ddtec.2017.11.007 |
| [20] |
M. Schmidt, A. Toplak, P.J.L.M. Quaedflieg, T. Nuijens, Curr. Opin. Chem. Biol. 38 (2017) 1-7. |
| [21] |
G.T. Debelouchina, T.W. Muir, Q. Rev. Biophys. 50 (2017) e7. DOI:10.1017/S0033583517000051 |
| [22] |
M. Rashidian, J.K. Dozier, M.D. Distefano, Bioconjugate Chem. 24 (2013) 1277-1294. DOI:10.1021/bc400102w |
| [23] |
J. Lotze, U. Reinhardt, O. Seitz, A.G. Beck-Sickinger, Mol. BioSyst. 12 (2016) 1731-1745. DOI:10.1039/C6MB00023A |
| [24] |
N. Stephanopoulos, M.B. Francis, Nat. Chem. Biol. 7 (2011) 876-884. DOI:10.1038/nchembio.720 |
| [25] |
Y. Yongsheng, X. Jiang, Sci. Chin. Chem. 59 (2016) 853-861. DOI:10.1007/s11426-016-5571-6 |
| [26] |
L.A. Marraffini, A.C. DeDent, O. Schneewind, Microbiol. Mol. Biol. Rev. 70 (2006) 192-221. DOI:10.1128/MMBR.70.1.192-221.2006 |
| [27] |
H. Ton-That, G. Liu, S.K. Mazmanian, K.F. Faull, O. Schneewind, Proc. Natl. Acad. Sci. U. S. A. 96 (1999) 12424-12429. DOI:10.1073/pnas.96.22.12424 |
| [28] |
O. Schneewind, A. Fowler, K.F. Faull, Science 268 (1995) 103-106. DOI:10.1126/science.7701329 |
| [29] |
S.K. Mazmanian, G. Liu, H. Ton-That, O. Schneewind, Science 285 (1999) 760-763. DOI:10.1126/science.285.5428.760 |
| [30] |
A.W. Maresso, O. Schneewind, Pharmacol. Rev. 60 (2008) 128-141. DOI:10.1124/pr.107.07110 |
| [31] |
S.K. Mazmanian, G. Liu, E.R. Jensen, E. Lenoy, O. Schneewind, Proc. Natl. Acad. Sci. U. S. A. 97 (2000) 5510-5515. DOI:10.1073/pnas.080520697 |
| [32] |
H. Mao, S.A. Hart, A. Schink, B.A. Pollok, J. Am. Chem. Soc. 126 (2004) 2670-2671. DOI:10.1021/ja039915e |
| [33] |
M.W. Popp, J.M. Antos, G.M. Grotenbreg, E. Spooner, H.L. Ploegh, Nat. Chem. Biol. 3 (2007) 707-708. DOI:10.1038/nchembio.2007.31 |
| [34] |
M.W.L. Popp, J.M. Antos, H.L. Ploegh, Curr. Protoc. Protein Sci. 56 (2009) 15.13. 11-15.13. 19. |
| [35] |
M.W.L. Popp, H.L. Ploegh, Angew. Chem. Int. Ed. 50 (2011) 5024-5032. DOI:10.1002/anie.v50.22 |
| [36] |
C.P. Guimaraes, M.D. Witte, C.S. Theile, et al., Nat. Protoc. 8 (2013) 1787-1799. DOI:10.1038/nprot.2013.101 |
| [37] |
R.L. Policarpo, H. Kang, X. Liao, et al., Angew. Chem. Int. Ed. 53 (2014) 9203-9208. DOI:10.1002/anie.201403582 |
| [38] |
X.L. Tan, M. Pan, Y. Zheng, et al., Chem. Sci. 8 (2017) 6881-6887. DOI:10.1039/C7SC02937C |
| [39] |
L. Chen, J. Cohen, X. Song, et al., Sci. Rep. 6 (2016) 31899. DOI:10.1038/srep31899 |
| [40] |
L.K. Swee, S. Lourido, G.W. Bell, J.R. Ingram, H.L. Ploegh, ACS Chem. Biol. 10 (2015) 460-465. DOI:10.1021/cb500462t |
| [41] |
X. Jia, S. Kwon, C.I.A. Wang, et al., J. Biol. Chem. 289 (2014) 6627-6638. DOI:10.1074/jbc.M113.539262 |
| [42] |
Z.M. Wu, S.Z. Liu, X.Z. Cheng, X.R. Zhao, H.F. Hong, Chin. Chem. Lett. 28 (2017) 553-557. DOI:10.1016/j.cclet.2016.11.001 |
| [43] |
J.M. Antos, M.C. Truttmann, H.L. Ploegh, Curr. Opin. Struct. Biol. 38 (2016) 111-118. DOI:10.1016/j.sbi.2016.05.021 |
| [44] |
L. Schmohl, D. Schwarzer, Curr. Opin. Chem. Biol. 22 (2014) 122-128. DOI:10.1016/j.cbpa.2014.09.020 |
| [45] |
X. Huang, A. Aulabaugh, W. Ding, et al., Biochemistry 42 (2003) 11307-11315. DOI:10.1021/bi034391g |
| [46] |
R. David Row, T.J. Roark, M.C. Philip, L.L. Perkins, J.M. Antos, Chem. Commun. 51 (2015) 12548-12551. DOI:10.1039/C5CC04657B |
| [47] |
I. Chen, B.M. Dorr, D.R. Liu, Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 11399-11404. DOI:10.1073/pnas.1101046108 |
| [48] |
H. Hirakawa, S. Ishikawa, T. Nagamune, Biotechnol. Bioeng. 109 (2012) 2955-2961. DOI:10.1002/bit.v109.12 |
| [49] |
H. Hirakawa, S. Ishikawa, T. Nagamune, J. Biotechnol. 10 (2015) 1487-1492. DOI:10.1002/biot.201500012 |
| [50] |
I. Wuethrich, J.G.C. Peeters, A.E.M. Blom, et al., PLoS ONE 9 (2014) e109883. DOI:10.1371/journal.pone.0109883 |
| [51] |
J. Shi, L. Kundrat, N. Pishesha, et al., Proc. Natl. Acad. Sci. U. S. A. 111 (2014) 10131-10136. DOI:10.1073/pnas.1409861111 |
| [52] |
J.M. Antos, J. Ingram, T. Fang, et al., Curr. Protoc. Protein Sci. 89 (2017) 15.13. 11-15.13. 19. |
| [53] |
S. Pritz, Y. Wolf, O. Kraetke, et al., J. Org. Chem. 72 (2007) 3909-3912. DOI:10.1021/jo062331l |
| [54] |
D.J. Williamson, M.A. Fascione, M.E. Webb, W.B. Turnbull, Angew. Chem. Int. Ed. 51 (2012) 9377-9380. DOI:10.1002/anie.201204538 |
| [55] |
D.J. Williamson, M.E. Webb, W.B. Turnbull, Nat. Protoc. 9 (2014) 253-262. DOI:10.1038/nprot.2014.003 |
| [56] |
F. Liu, E.Y. Luo, D.B. Flora, A.R. Mezo, J. Org. Chem. 79 (2014) 487-492. DOI:10.1021/jo4024914 |
| [57] |
Y.M. Li, Y.T. Li, M. Pan, et al., Angew. Chem. Int. Ed. 53 (2014) 2198-2202. DOI:10.1002/anie.201310010 |
| [58] |
J.M. Antos, G.L. Chew, C.P. Guimaraes, et al., J. Am. Chem. Soc. 131 (2009) 10800-10801. DOI:10.1021/ja902681k |
| [59] |
G.T. Hess, J.J. Cragnolini, M.W. Popp, et al., Bioconjugate Chem. 23 (2012) 1478-1487. DOI:10.1021/bc300130z |
| [60] |
G.K. Nguyen, S. Wang, Y. Qiu, et al., Nat. Chem. Biol. 10 (2014) 732-738. DOI:10.1038/nchembio.1586 |
| [61] |
G.K. Nguyen, A. Kam, S. Loo, et al., J. Am. Chem. Soc. 137 (2015) 15398-15401. DOI:10.1021/jacs.5b11014 |
| [62] |
G.K. Nguyen, Y. Qiu, Y. Cao, et al., Nat. Protoc. 11 (2016) 1977-1988. DOI:10.1038/nprot.2016.118 |
| [63] |
Y. Cao, G.K. Nguyen, J.P. Tam, C.F. Liu, Chem. Commun. 51 (2015) 17289-17292. DOI:10.1039/C5CC07227A |
| [64] |
X. Hemu, Y. Qiu, G.K. Nguyen, J.P. Tam, J. Am. Chem. Soc. 138 (2016) 6968-6971. DOI:10.1021/jacs.6b04310 |
| [65] |
X. Bi, J. Yin, G.K.T. Nguyen, et al., Angew. Chem. Int. Ed. 56 (2017) 7822-7825. DOI:10.1002/anie.201703317 |
| [66] |
K.S. Harris, T. Durek, Q. Kaas, et al., Nat. Commun. 6 (2015) 10199. DOI:10.1038/ncomms10199 |
| [67] |
R. Yang, Y.H. Wong, G.K.T. Nguyen, et al., J. Am. Chem. Soc. 139 (2017) 5351-5358. DOI:10.1021/jacs.6b12637 |
| [68] |
G.K. Nguyen, Y. Cao, W. Wang, C.F. Liu, J.P. Tam, Angew. Chem. Int. Ed. 54 (2015) 15694-15698. DOI:10.1002/anie.201506810 |
| [69] |
R.J. Siezen, J.A.M. Leunissen, Protein Sci. 6 (1997) 501-523. |
| [70] |
N. Vasantha, L.D. Thompson, C. Rhodes, et al., J. Bacteriol. 159 (1984) 811-819. |
| [71] |
L. Abrahmsen, J. Tom, J. Burnier, et al., Biochemistry 30 (1991) 4151-4159. DOI:10.1021/bi00231a007 |
| [72] |
T.K. Chang, D.Y. Jackson, J.P. Burnier, J.A. Wells, Proc. Natl. Acad. Sci. U. S. A. 91 (1994) 12544-12548. DOI:10.1073/pnas.91.26.12544 |
| [73] |
D.Y. Jackson, J. Burnier, C. Quan, M. Stanley, J. Tom, J.A. Wells, Science 266 (1994) 243-247. DOI:10.1126/science.7939659 |
| [74] |
A.C. Braisted, J.K. Judice, J.A. Wells, Methods Enzymol. 289 (1997) 298-313. DOI:10.1016/S0076-6879(97)89053-2 |
| [75] |
D.Y. Jackson, J.P. Burnier, J.A. Wells, J. Am. Chem. Soc. 117 (1995) 819-820. DOI:10.1021/ja00107a027 |
| [76] |
S. Atwell, J.A. Wells, Proc. Natl. Acad. Sci. U. S. A. 96 (1999) 9497-9502. DOI:10.1073/pnas.96.17.9497 |
| [77] |
A.M. Weeks, J.A. Wells, Nat. Chem. Biol. 14 (2017) 50. DOI:10.1038/nchembio.2521 |
| [78] |
S.H. Henager, N. Chu, Z. Chen, et al., Nat. Methods 13 (2016) 925-927. DOI:10.1038/nmeth.4004 |
| [79] |
A. Toplak, T. Nuijens, P.J.L.M. Quaedflieg, B. Wu, D.B. Janssen, Adv. Synth. Catal. 358 (2016) 2140-2147. DOI:10.1002/adsc.v358.13 |
| [80] |
T. Nuijens, A. Toplak, P.J.L.M. Quaedflieg, et al., Adv. Synth. Catal. 358 (2016) 4041-4048. DOI:10.1002/adsc.201600774 |
| [81] |
M. Schmidt, A. Toplak, H.J. Rozeboom, et al., Org. Biomol. Chem. 16 (2018) 609-618. DOI:10.1039/C7OB02812A |
| [82] |
M. Schmidt, A. Toplak, P.J.L.M. Quaedflieg, et al., Adv. Synth. Catal. 359 (2017) 2050-2055. DOI:10.1002/adsc.v359.12 |
| [83] |
M. Sunbul, J. Yin, Org. Biomol. Chem. 7 (2009) 3361-3371. DOI:10.1039/b908687k |
| [84] |
D. Roeser, A. Preusser-Kunze, B. Schmidt, et al., Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 81-86. DOI:10.1073/pnas.0507592102 |
| [85] |
B.L. Carlson, E.R. Ballister, E. Skordalakes, et al., J. Biol. Chem. 283 (2008) 20117-20125. DOI:10.1074/jbc.M800217200 |
| [86] |
K.V. Figura, B. Schmidt, T. Selmer, T. Dierks, Bioessays 20 (1998) 505-510. DOI:10.1002/(SICI)1521-1878(199806)20:6<>1.0.CO;2-P |
| [87] |
P. Bojarová, S.J. Williams, Curr. Opin. Chem. Biol. 12 (2008) 573-581. DOI:10.1016/j.cbpa.2008.06.018 |
| [88] |
I.S. Carrico, B.L. Carlson, C.R. Bertozzi, Nat. Chem. Biol. 3 (2007) 321. DOI:10.1038/nchembio878 |
| [89] |
P. Wu, W. Shui, B.L. Carlson, et al., Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 3000-3005. DOI:10.1073/pnas.0807820106 |
| [90] |
J.S. Rush, C.R. Bertozzi, J. Am. Chem. Soc. 130 (2008) 12240-12241. DOI:10.1021/ja804530w |
| [91] |
D. Rabuka, J.S. Rush, G.W. deHart, P. Wu, C.R. Bertozzi, Nat. Protoc. 7 (2012) 1052. DOI:10.1038/nprot.2012.045 |
| [92] |
E.M. Sletten, C.R. Bertozzi, Angew. Chem. Int. Ed. 48 (2009) 6974-6998. DOI:10.1002/anie.v48:38 |
| [93] |
E.-A. Kwak, J. Jaworski, J. Mater. Chem. B 1 (2013) 3486-3493. DOI:10.1039/c3tb20526f |
| [94] |
J.E. Hudak, H.H. Yu, C.R. Bertozzi, J. Am. Chem. Soc. 133 (2011) 16127-16135. DOI:10.1021/ja206023e |
| [95] |
R. M. Barfield, D. Rabuka, Leveraging formylglycine-generating enzyme for production of site-specifically modified bioconjugates, in: E. A. Lemke (Ed. ), Noncanonical Amino Acids: Methods and Protocols, Springer New York, New York, NY, 2018, pp. 3-16.
|
| [96] |
S.I. Liang, J.M. McFarland, D. Rabuka, Z.J. Gartner, J. Am. Chem. Soc. 136 (2014) 10850-10853. DOI:10.1021/ja504711n |
| [97] |
P.G. Holder, L.C. Jones, P.M. Drake, et al., J. Biol. Chem. 290 (2015) 15730-15745. DOI:10.1074/jbc.M115.652669 |
| [98] |
F.L. Zhang, P.J. Casey, Annu. Rev. Biochem. 65 (1996) 241-269. DOI:10.1146/annurev.bi.65.070196.001325 |
| [99] |
C.C. Palsuledesai, M.D. Distefano, ACS Chem. Biol. 10 (2015) 51-62. DOI:10.1021/cb500791f |
| [100] |
N. Kohl, S. Mosser, S. deSolms, et al., Science 260 (1993) 1934-1937. DOI:10.1126/science.8316833 |
| [101] |
J.B. Gibbs, A. Oliff, N.E. Kohl, Cell 77 (1994) 175-178. DOI:10.1016/0092-8674(94)90308-5 |
| [102] |
N.E. Kohl, F.R. Wilson, S.D. Mosser, et al., Proc. Natl. Acad. Sci. U. S. A. 91 (1994) 9141-9145. DOI:10.1073/pnas.91.19.9141 |
| [103] |
N. Berndt, A.D. Hamilton, S.M. Sebti, Nat. Rev. Cancer 11 (2011) 775-791. DOI:10.1038/nrc3151 |
| [104] |
U.T.T. Nguyen, J. Cramer, J. Gomis, et al., ChemBioChem 8 (2007) 408-423. DOI:10.1002/(ISSN)1439-7633 |
| [105] |
B.P. Duckworth, Z. Zhang, A. Hosokawa, M.D. Distefano, ChemBioChem 8 (2007) 98-105. DOI:10.1002/(ISSN)1439-7633 |
| [106] |
M. Rashidian, J.K. Dozier, S. Lenevich, M.D. Distefano, Chem. Commun. 46 (2010) 8998-9000. DOI:10.1039/c0cc03305g |
| [107] |
G.R. Labadie, R. Viswanathan, C.D. Poulter, J. Org. Chem. 72 (2007) 9291-9297. DOI:10.1021/jo7017747 |
| [108] |
C. Gauchet, G.R. Labadie, C.D. Poulter, J. Am. Chem. Soc. 128 (2006) 9274-9275. DOI:10.1021/ja061131o |
| [109] |
B.P. Duckworth, J. Xu, T.A. Taton, A. Guo, M.D. Distefano, Bioconjugate Chem. 17 (2006) 967-974. DOI:10.1021/bc060125e |
| [110] |
B.E. Dursina, R. Reents, A. Niculae, et al., Protein Expres. Purif. 39 (2005) 71-81. DOI:10.1016/j.pep.2004.09.015 |
| [111] |
J.K. Dozier, S.L. Khatwani, J.W. Wollack, et al., Bioconjugate Chem. 25 (2014) 1203-1212. DOI:10.1021/bc500240p |
| [112] |
M. Fernández-Suárez, H. Baruah, L. Martínez-Hernández, et al., Nat. Biotechnol. 25 (2007) 1483. DOI:10.1038/nbt1355 |
| [113] |
S. Puthenveetil, D.S. Liu, K.A. White, S. Thompson, A.Y. Ting, J. Am. Chem. Soc. 131 (2009) 16430-16438. DOI:10.1021/ja904596f |
| [114] |
I. Chen, M. Howarth, W. Lin, A.Y. Ting, Nat. Methods 2 (2005) 99. DOI:10.1038/nmeth735 |
| [115] |
M. Howarth, A.Y. Ting, Nat. Protoc. 3 (2008) 534. DOI:10.1038/nprot.2008.20 |
| [116] |
D. Schumacher, J. Helma, F.A. Mann, et al., Angew. Chem. Int. Ed. 54 (2015) 13787-13791. DOI:10.1002/anie.201505456 |
| [117] |
C.W. Lin, A.Y. Ting, J. Am. Chem. Soc. 128 (2006) 4542-4543. DOI:10.1021/ja0604111 |
| [118] |
P. Dennler, A. Chiotellis, E. Fischer, et al., Bioconjugate Chem. 25 (2014) 569-578. DOI:10.1021/bc400574z |
| [119] |
W.P. Heal, S.R. Wickramasinghe, P.W. Bowyer, et al., Chem. Commun. 48 (2008) 480-482. |