 2018, Vol. 29
2018, Vol. 29 



b The Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem 91904, Israel;
c State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China
In the past decades, peptides and proteins have found broad applications in pharmaceutical, vaccination, nutrition, and other life sciences. As a consequence, large amountsofpure andhomogeneous peptides andproteins areinhighlydemand, and thus stimulating the development of peptide synthetic methodology. Since 1882, when Curtius constructed the first peptide bond [1], chemical synthesis of peptide has witnessed a significant development with several milestone breakthroughs [2-4]. In 1932, Bergmann introduced an easily removable amino-protecting group, the carbobenzoxy (Cbz) group [5], which couldprotect the aminofunction temporarily, led to a new era in peptide synthesis. In 1954, Du Vigneaud accomplished the chemical total synthesis of the octapeptide oxytocin, the first manmade peptide hormone possessing the same biological activity as that isolated from nature [6]. Du Vigneaudwas awarded the Noble prize in chemistry in 1955, and this event stimulates the chemical synthesis of proteins and spurs the development of coupling reagents and racemization suppressors [7]. The most important breakthrough in this field is the solid-phase peptide synthesis (SPPS) [8, 9], invented by Merrifield in1963.His "simple and ingenious" idea reduced the time required for peptide synthesis from years to days and made many once-unthinkable syntheses possible. Twenty years later, the Noble prize in chemistry was awarded to Merrifield to honorhisimmense contribution topeptide synthesis.Thanksfor this technology, peptides with 40–50 amino acids could be synthesized smoothly by SPPS even automatically by using commercial available peptide synthesizer. However, the intrinsic limitation of SPPS complicates purification and decrease yield when it comes to long peptide sequence. The straightforward strategy for the synthesis of large peptide and protein is to join short peptide segments together via a native peptide bond [10-12]. A range of methods including prior thiol capture [13-16], native chemical ligation (NCL) [17, 18], sugar-assisted glycopeptide ligations (SAL) [19, 20], traceless staudinger ligation [21-23], ketoacid–hydroxylamine ligation (KAHA) [24-26], serine/threonine ligation (STL) [27-33] have been developed to facilitate peptide segment condensation in past decades. Among them, NCL invented by Kent in 1994 is the most successful one and has been widely used in chemical synthesis of proteins [17, 18]. This strategy featured with the reaction of peptide C-terminal thioester with N-terminal cysteine peptide under mild reaction conditions. Enormous efforts have been dedicated to NCL by developing novel synthetic methods for peptide thioester [34-40] and temporarily introducing of a removable mercapto group to other nature amino acids [41-45] to expand its application in peptide and protein chemical biology. Despite great progress in chemical peptide ligation has been made over the last century, chemical synthesis of large proteins still be a tremendous challenge for chemical biologists [46-51].
In this regard, the enzyme-mediated peptide ligation offers great advantages over chemical procedures [52-54]. Two reaction mechanisms are involved in enzyme-mediated peptide ligations. One is the direct reverse reaction of protease-catalyzed peptide hydrolysis (i.e., thermodynamically controlled process, Fig. 1a) [55, 56]. The other is the aminolysis of N-protected peptide esters (i.e., kinetically controlled approach, Fig. 1b) [57]. Despite all of the proteases are amenable for the thermodynamic approach, high enzyme loading and elaborate manipulation of reaction media are necessary to guarantee a moderate velocity. In contrast, kineticcontrolled strategy has a faster velocity and low enzyme requirement. Nevertheless, beside the competition of hydrolysis, ester substrates are required by kinetic approach [58]. We refer the readers to other excellent reviews for more comprehensive discussions about the reaction mechanisms of enzyme-mediated peptide ligation [52, 57-60]. In fact, enzyme-mediated peptide synthesis is about as old as chemical peptide synthesis. Enzyme catalyzed peptide synthesis was first predicted by van't Hoff in 1898 [61], and was realized by Bergmann in 1938 [55]. However, enzyme-mediated peptide synthesis has been impeded by the difficult availability of peptide ligases. Fortunately, with the fast development of biological technology, including genetic mining, protein engineering, and recombinant DNA technologies, the pace of discovery and optimization of peptide ligases has accelerated, and thus stimulates the renaissance of enzyme-mediated peptide and protein synthesis [54, 59, 62-72]. Herein, we briefly summarized the recent advances in the enzyme-mediated peptide ligation and highlight their application in protein syntheses, labelling and site-specific modifications.

|
Download:
|
| Fig. 1. Reaction mechanisms of enzyme-mediated peptide ligation | |
{kind=link}
2. Peptide ligase from nature
Enzymes, that mediate peptide and protein hydrolysis, also known as proteases are abundantly available from nature. On the contrary, peptide ligases, enzymes that catalyze the peptide bond formation are very rare. Even so, chemical biologists have never stopped looking for efficient peptide ligase from various living forms because of the advantages of enzyme-mediated peptide ligation such as mild reaction conditions, high regioselectivity, small toxicity, racemization-free. Thanks for these long-standing efforts, a few peptide ligases have been discovered from nature.
2.1. Sortase ASortase A isolated from Staphylococcus aureus [73, 74] is the most widely used peptide ligase in the chemoselective ligations and modifications of peptides and proteins [75-78]. In the presence of Ca2+, sortase A cleaves amide bond between the Thr and Gly of a conserved C-terminal LPXTG motif (X = any amino acid), which is the sortase recognition sequence in the target protein, to form an acyl enzyme intermediate. Then this intermediate is attacked by an aminoglycine nucleophile to generate the desired ligation product (Fig. 2). Owning to its robustness and easy availability by recombinant technology, sortase A has been widely used to mediate conjugation of protein-peptide or protein-protein in various research fields of chemical biology.

|
Download:
|
| Fig. 2. Sortase-mediated peptide ligation | |
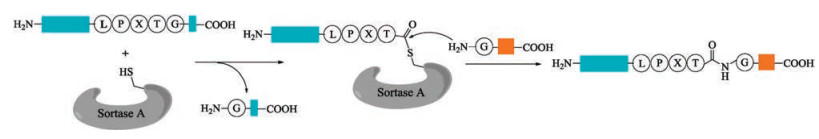{kind=link}
In 2004, Mao and co-workers added the sorting motif to the Cterminus of recombinant green fluorescent protein (GFP), which was subsequently ligated with various glycine-containing nucleophiles with the catalysis of bacterial sortase [79]. This is a robust method for site-specific protein modification with a wide range of functional peptides. Based on this concept, a series of applications of sortase-mediated ligation (SML) such as fluorescence labeling of fusion proteins [80], modification of proteins on the surface of phage particles with small molecules (fluorophores, biotin) has been developed (Fig. 3) [81]. Notably, modification of antibodyproteins to optimize antigen presentation [82-84], protein cyclization to increase rigidity and resistance to enzymatic degradation [85-87], and protein immobilization can also be accomplished by using the sortase-mediated transpeptidation strategy [88].

|
Download:
|
| Fig. 3. Applications of sortase-mediated peptide ligations | |
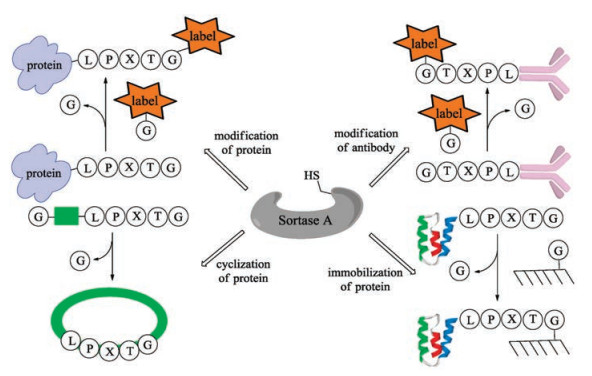{kind=link}
In recent years, SML has been one of the most attractive ligation technologies in biological chemistry. However, the shortcomings of SML create the limitation for application in much wider fields. The poor catalytic efficiency of sortase A requires extremely high enzyme concentration and extended coupling time are necessary to complete the reaction. Another problem is the reversibility of the ligation reactions which limits the ligation yield to approximately 50% by using equal concentrations of starting material. To make the equilibrium move toward to the desired product side, large excess of one of the reactants or timely removing of the byproducts is required [89, 90]. Other strategies to address this problem include the elaborate designing of either transpeptidation products and resorting to other sorting motifs. The ligation product can be engineered to form an unreactive β-hairpin at sortase recognition site, which inhibits the access of sortase to the reassemble sorting motif [91, 92]. The development of the sorting motif has also been achieved by releasing non-nucleophilic leaving group. The modified sorting motif with a methylester replaced the C-terminal glycine residue is reported by Ploegh and co-workers [93], the relative unreactive methanol was released with this substrate during the process of ligation (Fig. 4a). However, excess amounts of enzymes and methylester are required to guarantee a good yield. Later, Turnbull and co-workers optimized this method by replacing the C-terminal glycine with a glycolic acid [94], which was processed much faster than LPXT-OMe substrates with equal concentrations of ligation partners (Fig. 4b). Furthermore, Liu and co-workers designed a smart sorting motif with a depsipeptide, which can generate a diketopiperazine cyclic dipeptide spontaneously upon release (Fig. 4c) [95]. In 2014, Liu and co-workers reported a novel tactic to prepare recombinant protein hydrazides by taking advantage of the irreversible feature of sortase-mediated hydrazinolysis of proteins (Fig. 4d) [96]. This strategy was further applied to synthesis of non-natural cyclic analogues of antibacterial peptides P-113 by Wu and co-workers [97]. All of these methods represent valuable alternatives to perform the sotasemediated ligation reaction irreversibly.

|
Download:
|
| Fig. 4. Sortase-mediated irreversible peptide ligations | |
{kind=link}
2.2. Butelase 1
The long recognition motif (LPXTG) of sotase A restricts its application in certain proteins containing such sequence. Recently, this disadvantage was partially overcome by another natural peptide ligase, butelase 1, with a more flexible ligation site. Butelase 1 is a natural ribosomal peptide ligase involved in the biosynthesis of cyclotides in a tropical medicinal plant C. ternatea, which was isolated by Tam and co-workers in 2014 [98]. It is a promising alternative to sortase. It selectively recognizes a Asx-His-Val tripeptide motif and cleaves the peptide bond between Asx and His to ligate with N-terminal residue (Xaa) to form a new Asx-Xaa bond and thus affording a traceless peptide ligation (Fig. 5). Compared to previous peptide ligases with a Kcat of 1 h-1 or 1 d-1, butelase 1 is much more efficient with a Kcat of 17 s-1. In addition, it has an extremely high catalytic efficiency (only approx. 0.005 molar equivalents of enzyme was required). Although butelase 1 has a 71% sequence identity and the same catalytic triad with that of legumain, butelase 1 shows the ligation ability while not hydrolyzes the substrate of legumain. For most amino acids at the "P1" position of N-terminal, butelase 1 catalyzes efficiently peptide ligation, except for Pro, Asp and Glu. The hydrophobic amino acids such as Ile, Leu and Val at the "P2" position display a better performance.

|
Download:
|
| Fig. 5. Butelase-mediated peptide ligations | |
{kind=link}
Since the high efficiency of butelase 1, a series of applications of butelase-mediated ligations (BML) has been reported (Fig. 6). For example, butelase-mediated cyclization (BMC) of GFP and human growth hormone (somatropin) were completed in 15 min with >95% yields [99]. In addition, the highly hydrophobic circular bacteriocins, the largest antimicrobial peptides, were synthesized successfully by using BMC as a key step [100]. Notably, BMC is also amenable to the macrocyclization of D-amino acids containing peptide [101]. In 2015, Liu and Tam developed a novel tactics to prepare protein thioesters through BML of target protein with GlyCOSR [102]. It is worth mentioning that this method could be combined with NCL and sortase-mediated ligation (SML) in tandem for protein labeling. To further exploit BML's powerful compatibility, Liu and Tam reported a novel enzymatic modification of live E. coli cell with diverse molecules (fluorophores, biotin, glycosylated peptide) [103]. Despite a short time after its discovery, butelase 1 has already exhibited its potential applicability in site-specific modification and synthesis of peptides and proteins and enormous potential in fundamental biomedical research.

|
Download:
|
| Fig. 6. Applications of butelase-mediated ligations | |
{kind=link}
Although butelase 1 is the fastest known peptide ligase, like any other methods, BML has its own intrinsic limitations. Similar with that of sortase A, BML is also reversible, and excess amount of substrate is required to ensure completion of the reaction. To overcome this drawback, Tam and Liu developed an irreversible intermolecular peptide ligation by employing thiodepsipeptide as the sorting signal. For model peptide and ubiquitin, quantitative ligation yields could be achieved with a small excess of substrate [104]. In addition, peptide dendrimers have been prepared successfully through the butelase-mediated bioconjugation of thiodepsipeptides with lysyl dendrimeric scaffolds [105]. However, recombinant expression of butelase 1 with equivalent functional activity has not yet been achieved [106], limiting its potential in biotechnological applications. It is expected that this issue will be overcome in the future with the development of biological technology.
2.3. Ligases for peptide cyclizationCyanobactins are ribosomal peptides produced from various cyanobacteria, and further extensively modified by enzymes to produce a variety of cyclic natural products [107]. Inspired by this phenomenon, Schmidt and coworkers discovered a protease PatG, a peptide cyclase expressed from a cyanobactin gene cluster, which was cloned from the metagenome of a coral reef animal [108]. Flanked recognition motif of AYDGE was cleaved to enable a traceless tandem cyclization of R-AYDGE (R = peptide) (Fig. 7a). Later, Covello and coworkers discovered two enzymes-oligopeptidase 1 (OLP1) and cyclase (PCY1) [109], which were extracted and purified from a plant named Saponaria vaccaria (Caryophyllaceae). OLP1 cleaves the precursor peptide (presegetalin A1) at the Nterminal of 32-amino acid linear peptide, and then PCY1 cyclizes the linear peptide intermediate to offer the cyclic peptide segetalin A (6-amino acid) with concurrent removal of a C-terminal flanking sequence (Fig. 7b). One year later, Walton and coworkers extracted a specific prolyl oligopeptidase (GmPOPB) from Galerina marginata [110], which selectively recognizes proline from the propeptide. First, it catalyzes GmAMA1 (a 35-amino acid propeptide) hydrolysis at an internal Pro to furnish the C-terminal 25-mer product, then transpeptidation at the second Pro produces the cyclooctapeptide (Fig. 7c). All of these three cyclases are serine proteases and require a propeptide. Compared to butelase, PatG and PCY1 exhibit slow kinetics with a Kcat of 1 h-1 or 1 d-1, while GmPOPB has comparable turnover rates and similar Km values.

|
Download:
|
| Fig. 7. Ligase-mediated peptide cyclization | |
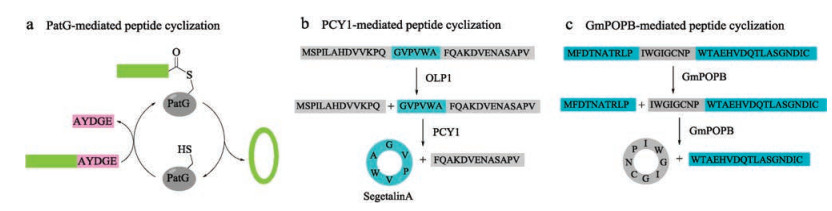{kind=link}
3. Artificial peptide ligase via protein engineering
Because of the rare occurrence of peptide ligase in nature, scientists resort to artificial enzymes by employing protein engineering technologies. Inspired by the reversible character of protease catalyzed hydrolysis of peptides and proteins, many efforts have been made to reverse the hydrolysis functionality of a protease to furnish a ligase. Three strategies have been employed to transfer a protease to a ligase. First, the hydrophilic active-site (Cys) is mutated to the hydrophobic amino acid (Ala) to prohibit the approach of water molecular to RCOE and thus suppressing the hydrolysis of the RCOE (Fig. 1b) [111]. Secondly, the catalytic residue Ser is converted to a Cys due to the inherently greater lability of thioesters toward amines compared to water [112]. Thirdly, amino acids with large steric hindrance near the active site are converted to small sized ones to favor amines entering the catalytic patch, and thus resulting a faster aminolysis of RCOE [113]. Extensive studies have been carried out to optimize the structure of the low efficient natural peptide ligases to improve their ligation efficiency.
3.1. Sortase A variantsSortagging is a powerful strategy for the site-specific labeling of peptides and proteins under mild reaction conditions, but have some limitations such as narrow substrate scope, poor reaction rates and a dependence on a Ca2+ cofactor. To address these shortcomings, a number of optimizations have been performed. In 2011, Schwarzer and co-workers identified that F40-sortase, a mutant of sortase A, possesses a broader selectivity profile regarding position 1 of the sorting motif (Ala, Asp, Ser, Pro, Gly) [114]. Roy and Choudhury delineated the conformational signatures of a catalytically competent substrate in sortase A using substrate analogues containing nonproteinogenic amino acids [115]. In 2017, Antos and co-workers developed several sortase homologs to expand the substrate scope of sortase-mediated ligations [116]. Chen and co-workers integrated five underlying mutations (P94R/D160 N/D165 A/ K190E/K196 T) into a single gene to get a sortase A mutant, kcat of which was approximately 120 times higher than that of sortase A [117]. Subsequently, Hirakawa and co-workers further enhanced the catalytic activity of sortase A by introducing two additional mutations (E105 K/E108 A or E105 K/ E108Q) [118]. In addition, these variants eliminate the Ca2+ dependence. In 2016, a new strategy of in vitro compartmentalization was developed to identify new Ca2+ independent sortase A mutants [119]. Compared with wild type sortase A (in presence of Ca2+), this variant displayed slight improvement in the absence of Ca2+. Undoubtedly, these efforts will broaden the applications of sortase A remarkably.
3.2. OaAEP1 variantsOaAEP1 is a protease isolated from Oldenlandia affinis [120]. It displays a slight peptide cyclase activity, and shares 66% sequence identity with that of butelase 1. With a special recognition sequence of Asx-Gly-Leu (Fig. 8), OaAEP1 has a lower catalytic activity compared with butelase 1. The advantage OaAEP1 over butelase 1 is that OaAEP1 can be obtained by recombinant expression. The structure of OaAEP1 was established by X-ray crystallography analysis by Wu and co-workers [111]. Based on the structure, the catalytic active site was identified and thus offering an opportunity for further optimization. In fact, the catalytic activity of OaAEP1 was further enhanced via protein engineering. Wu and co-workers identified the Kcat of a mutant was 160 times higher than that of wild type OaAEP1 via mutation of Cys247Ala.

|
Download:
|
| Fig. 8. OaAEP 1 mediated peptide ligations | |
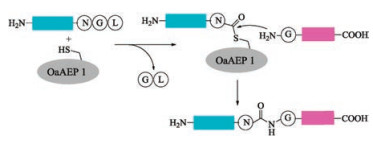{kind=link}
3.3. Trypsin variants
Trypsin and trypsin variants catalyzed peptide synthesis has been known for decades [121]. In 2014, Bordusa and coworkers described a variant of trypsin, termed trypsiligase, for the sitespecific labeling of both N- and C-terminal of target peptide or protein in one-pot manner [122]. This enzyme displays high selective specificity for the tripeptide (YRH) recognition sequence in the presence of Zn2+ and the cleavage of the peptide bond between Tyr and Arg allows for the pre-installation of desired tag to the target proteins. Importantly, the mode of action of trypsiligase provided the first example of substrate-activated catalysis, which is active exclusively only in the presence of appropriate substrates. What is more, trypsiligase recognizes the substrate mimetic of peptidyl 4-guanidinophenyl esters (OGp), resulting in kinetic favoring of the amidolysis during the modification of N-terminal of RH-proteins (Fig. 9a) [123]. Trypsiligase-mediated C-terminal modifications of protein could also be realized by introducing Y-RH recognition sequence to Cterminal with a RH-X (X = peptide, tag) peptide as an acyl acceptor (Fig. 9b) [124]. Trypsiligase-mediated ligation reaction is much quickly (minutes), and requires approximately 0.1 molar equivalents of enzyme. However, an excess of acyl acceptor substrate (often 10 equiv.) is necessary. In addition, the tripeptide recognition sequence is found in only 0.5% of all known protein sequences in the SwissProt database [122], which restricts its application in protein synthesis. Moreover, back reactions and hydrolysis take place in the presence of YRH-containing substrates during the Cterminal protein modifications. The widespread application of trypsiligase remains a challenge, until these drawbacks be solved.

|
Download:
|
| Fig. 9. Trypsiligase-mediated N- and C-terminal modification of protein | |
{kind=link}
3.4. Subtilisin variants
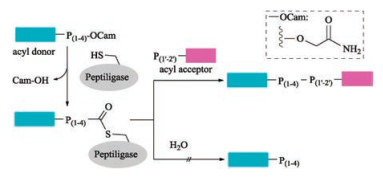
Subtilisin isolated from Bacillus amyloliquefaciens is a serine protease with broad substrate specificity. Its differentness from the chymotrypsin-trypsin proteases family made it a popular target peptide ligase for modification [125, 126]. Thirty years ago, Kaiser and coworkers reported a method for peptide segment coupling catalyzed by thiolsubtilisin, a subtilisin variant in which the activesite Ser221 was replaced with Cys (S221C). Upon such a manipulation, the ability of thiolsubtilisin to catalyze peptide ligation was improved by >1000-fold compared with that of hydrolysis [127]. Shortly after that, a more powerful subtilisin variant, subtiligase was designed by Wells with a double mutation (S221C, P225 A) [112, 128]. Although subtiligase exhibits a higher peptide ligase activity relative to thiolsubtilisin, an excess amount of acyl acceptor is still required to suppress substantial amounts of hydrolysis. Further optimization of subtilisin was continued and resulted a new variant by mutating Tyr to Lys (Y217 K) [129]. With the catalysis of such subtiligase, the ligation of Cys-free peptides to protein thioesters was performed successfully with good conversion efficiency. Almost at the same time, Janssen and Wu reported another novel Ca2+-independent subtilisin variant termed peptiligase (Fig. 10) [130], which proceed via attacking the activated Cterminal carboxamido-methyl (Cam)-ester fragment by acylacceptor nucleophilic with insignificant amounts of hydrolysis. Notably, peptiligase-mediated ligations are not reversible in aqueous media, leading to the high average ligation yields (up to 98% in < 1 h) by using 1.1–1.5 equivalents of acyl acceptor. Furthermore, head-to-tail macrocyclizations of various linear peptides can be achieved by peptiligase. Peptiligase has a recognition motif of six amino acid sequence, four of which are involved in acyl donor (S1–S4) and two are involved in acyl acceptor (S1' and S2'). S1' was found to be highly discriminative and only small amino acids such as serine, glycine and alanine are accepted at this position. Additionally, proline should be avoided in both S1 and S2' positions. Only hydrophobic or slightly polar amino acids are tolerated at S4 position. Later, they reported a broader applicable and high catalytic efficient peptiligase variant termed omniligase-1 [113], which has broad substrate scope of S1' site except for proline. Gram-scale synthesis of exenatide and Nterminal modification of human serum albumin with exenatide was successfully achieved by employing omniligase-1-mediated peptide ligation. Thymosin-α1, an acetylated therapeutic peptide contains 28 amino acid for treatment of hepatitis B and C [131, 132], is difficult to be synthesized via conventional chemical methods. Recently, Nuijens and coworkers reported that thymoligase, another peptiligase variant, is effective for Thymosin-α1 synthesis by ligating 14-mer plus 14-mer peptide segments with a yield of two-fold higher than those typical industrial processes [133].

|
Download:
|
| Fig. 10. Peptiligase-mediated peptide ligations | |
{kind=link}
Aimed at providing peptide ligase with a flexible recognition motif, Wells and coworkers identified a range of 72 subtiligase mutants that can site-selective modify N-terminal sequences, which were extremely difficult to modify [134]. The comprehensive survey of subtiligases specificity was achieved by using α-amine acceptor peptide libraries generated from digested E. coli proteome and analyzing the frequency of each amino acid in each position. With this technology, termed proteomic identification of ligation sites (PILS), they investigated the ligation efficiency for over 25, 000 enzyme-substrate pairs for the engineered subtiligase variants. The modification of various peptides and proteins also proceeded smoothly by employing the newly identified ligases. This work provides critical detail information to develop strategies for finding the best ligase [135].
4. Summary and perspectiveThe manipulation of peptide and protein plays an important role in peptide drug discovery, protein engineering and other life sciences, and technologies developed in this area is becoming an active field of research. Owing to the mild reaction conditions, racemization-free at the ligation site, no need for protection of side chain functional groups, compatible with native protein substrates and amenable for scale-up reactions, enzyme-catalyzed peptide ligation has evolved into a valuable alternative for chemical approach. Although finding a universally applicable peptide ligase is impossible because of the diverse nature of proteins, the difficult situation of searching for peptide ligase with relatively broad flexibility of recognition sequences was alleviated by modern biological technologies. With the continuous innovation of biological technology and the deepening of people's understanding, the restriction of peptide ligases will be overcome in the future. As the potential of peptide ligase may well exceed that of chemical methods, a renaissance of enzyme-mediated peptide ligation is coming.
AcknowledgmentsThe National Natural Science Foundation of China (Nos. 21462023, 21778025) and the Education Department of Jiangxi Province (No. 150297) are acknowledged for support of this research.
| [1] |
T. Curtius, J. Prakt, Chemie 26 (1882) 145-208. |
| [2] |
V.R. Pattabiraman, J.W. Bode, Nature 480 (2011) 471-479. DOI:10.1038/nature10702 |
| [3] |
C.P. Hackenberger, D. Schwarzer, Angew. Chem. Int. Ed. 47 (2008) 10030-10074. DOI:10.1002/anie.v47:52 |
| [4] |
T. Kimmerlin, D. Seebach, Chem. Biol. Drug Des. 65 (2005) 229-260. |
| [5] |
M. Bergmann, L. Zervas, Ber. Deut. Chem. Ges. 65 (1932) 1192-1201. DOI:10.1002/cber.19320650722 |
| [6] |
V. du Vigneaud, C. Ressler, J.M. Swan, et al., J. Am. Chem. Soc. 76 (1954) 3115-3121. DOI:10.1021/ja01641a004 |
| [7] |
A. El-Faham, F. Albericio, Chem. Rev. 111 (2011) 6557-6602. DOI:10.1021/cr100048w |
| [8] |
R.B. Merrifield, J. Am. Chem. Soc. 85 (1963) 2149-2154. DOI:10.1021/ja00897a025 |
| [9] |
R.B. Merrifield, Angew. Chem. Int. Ed. 24 (1985) 799-810. DOI:10.1002/(ISSN)1521-3773 |
| [10] |
C.P. Hackenberger, D. Schwarzer, Angew. Chem. Int. Ed. 47 (2008) 10030-10074. DOI:10.1002/anie.v47:52 |
| [11] |
V.R. Pattabiraman, J.W. Bode, Nature 480 (2011) 471-479. DOI:10.1038/nature10702 |
| [12] |
J.H. Yang, J.F. Zhao, Sci. China Chem. 61 (2018) 97-112. DOI:10.1007/s11426-017-9056-5 |
| [13] |
D.S. Kemp, S.-L. Leung, D.J. Kerkman, Tetrahedron Lett. 22 (1981) 181-184. DOI:10.1016/0040-4039(81)80049-4 |
| [14] |
D.S. Kemp, D.J. Kerkman, Tetrahedron Lett. 22 (1981) 185-186. DOI:10.1016/0040-4039(81)80050-0 |
| [15] |
D.S. Kemp, N.G. Galakatos, B. Bowen, et al., J. Org. Chem. 51 (1986) 1829-1838. DOI:10.1021/jo00360a033 |
| [16] |
D.S. Kemp, R.I. Carey, J. Org. Chem. 58 (1993) 2216-2222. DOI:10.1021/jo00060a043 |
| [17] |
P.E. Dawson, T.W. Muir, l. Clarklewis, et al., Science 266 (1994) 776-779. DOI:10.1126/science.7973629 |
| [18] |
S.B.H. Kent, Chem. Soc. Rev. 38 (2009) 338-351. DOI:10.1039/B700141J |
| [19] |
A. Brik, C.-H. Wong, Chem. Eur. J. 13 (2007) 5670-5675. DOI:10.1002/(ISSN)1521-3765 |
| [20] |
R.J. Payne, S. Ficht, W.A. Greenberg, et al., Angew. Chem. Int. Ed. 47 (2008) 4411-4415. DOI:10.1002/(ISSN)1521-3773 |
| [21] |
M. Köhn, R. Breinbauer, Angew. Chem. Int. Ed. 43 (2004) 3106-3116. DOI:10.1002/(ISSN)1521-3773 |
| [22] |
B.L. Nilsson, L.L. Kiessling, R.T. Raines, Org. Lett. 2 (2000) 1939-1941. DOI:10.1021/ol0060174 |
| [23] |
E. Saxon, C.R. Bertozzi, Science 287 (2000) 2007-2010. DOI:10.1126/science.287.5460.2007 |
| [24] |
J.W. Bode, R.M. Fox, K.D. Baucom, Angew. Chem. Int. Ed. 45 (2006) 1248-1252. DOI:10.1002/(ISSN)1521-3773 |
| [25] |
I. Pusterla, J.W. Bode, Angew. Chem. Int. Ed. 51 (2012) 513-516. DOI:10.1002/anie.201107198 |
| [26] |
I. Pusterla, J.W. Bode, Nat. Chem. 7 (2015) 668-672. DOI:10.1038/nchem.2282 |
| [27] |
X. Li, H.Y. Lam, Y. Zhang, et al., Org. Lett. 12 (2010) 1724-1727. DOI:10.1021/ol1003109 |
| [28] |
Y. Zhang, C. Xu, H.Y. Lam, et al., Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 6657-6662. DOI:10.1073/pnas.1221012110 |
| [29] |
C.L. Lee, H. Liu, C.T.T. Wong, et al., J. Am. Chem. Soc. 138 (2016) 10477-10484. DOI:10.1021/jacs.6b04238 |
| [30] |
C.F. Liu, J.P. Tam, Proc. Natl. Acad. Sci. U. S. A. 91 (1994) 6584-6588. DOI:10.1073/pnas.91.14.6584 |
| [31] |
C.F. Liu, J.P. Tam, J. Am. Chem. Soc. 116 (1994) 4149-4153. DOI:10.1021/ja00089a001 |
| [32] |
C.L. Lee, X.C. Li, Sci. Chin. Chem. 59 (2016) 1061-1064. |
| [33] |
C. Xu, J. Xu, H. Liu, et al., Chin. Chem. Lett. 29 (2018) 1113-1116. DOI:10.1016/j.cclet.2018.05.012 |
| [34] |
F. Mende, O. Seitz, Angew. Chem. Int. Ed. 50 (2011) 1232-1240. DOI:10.1002/anie.v50.6 |
| [35] |
J.S. Zheng, S. Tang, Y.C. Huang, et al., Acc. Chem. Res. 46 (2013) 2475-2484. DOI:10.1021/ar400012w |
| [36] |
J.S. Zheng, H.N. Chang, F.L. Wang, et al., J. Am. Chem. Soc. 133 (2011) 11080-11083. DOI:10.1021/ja204088a |
| [37] |
G.M. Fang, J.X. Wang, L. Liu, Angew. Chem. Int. Ed. 51 (2012) 10347-10350. DOI:10.1002/anie.201203843 |
| [38] |
H.X. Li, S.W. Dong, Sci. China. Chem. 60 (2017) 201-213. |
| [39] |
G.M. Fang, J.X. Wang, L. Liu, Angew. Chem. Int. Ed. 51 (2012) 10347-10350. DOI:10.1002/anie.201203843 |
| [40] |
J.S. Zheng, S. Tang, Y.K. Qi, et al., Nat. Protoc. 8 (2013) 2483-2495. DOI:10.1038/nprot.2013.152 |
| [41] |
H. Rohde, O. Seitz, Biopolymers 94 (2010) 551-559. DOI:10.1002/bip.21442 |
| [42] |
J. Ma, J. Zeng, Q. Wan, Top. Curr. Chem. 363 (2015) 57-101. |
| [43] |
L.Z. Yan, P.E. Dawson, J. Am. Chem. Soc. 123 (2001) 526-533. DOI:10.1021/ja003265m |
| [44] |
Q. Wan, S.J. Danishefsky, Angew. Chem. Int. Ed. 46 (2007) 9248-9252. DOI:10.1002/(ISSN)1521-3773 |
| [45] |
Q.Q. He, G.M. Fang, L. Liu, Chin. Chem. Lett. 24 (2013) 265-269. DOI:10.1016/j.cclet.2013.03.013 |
| [46] |
A.A. Vinogradov, E.D. Evans, B.L. Pentelute, Chem. Sci. 6 (2015) 2997-3002. DOI:10.1039/C4SC03877K |
| [47] |
S. Tang, Y.Y. Si, Z.P. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 5713-5717. DOI:10.1002/anie.201500051 |
| [48] |
M. Pan, S. Gao, Y. Zheng, et al., J. Am. Chem. Soc. 138 (2016) 7429-7435. DOI:10.1021/jacs.6b04031 |
| [49] |
M. Jbara, S.K. Maity, M. Morgan, et al., Angew. Chem. Int. Ed. 55 (2016) 4972-4976. DOI:10.1002/anie.v55.16 |
| [50] |
Z. Wang, W. Xu, L. Liu, et al., Nat. Chem. 8 (2016) 698-704. DOI:10.1038/nchem.2517 |
| [51] |
A.M. Levinson, J.H. McGee, A.G. Roberts, et al., J. Am. Chem. Soc. 139 (2017) 7632-7639. DOI:10.1021/jacs.7b02988 |
| [52] |
J. Bongers, E.P. Heimer, Peptides 15 (1994) 183-193. DOI:10.1016/0196-9781(94)90189-9 |
| [53] |
F. Guzman, S. Barberis, A. Illanes, Electron. J. Biotechnol. 10 (2007) 279-314. |
| [54] |
M. Schmidt, A. Toplak, P.J. Quaedflieg, et al., Curr. Opin. Chem. Biol. 38 (2017) 1-7. |
| [55] |
M. Bergmann, H.F. Conrat, D.G. Doherty, J. Biol. Chem. 124 (1938) 1-6. |
| [56] |
M. Bergmann, H.F. Conrat, J. Biol. Chem. 119 (1937) 707-720. |
| [57] |
C.H. Wong, K.T. Wang, Experientia 47 (1991) 1123-1129. DOI:10.1007/BF01918376 |
| [58] |
H.D. Jakubke, P. Kuhl, A. Könnecke, Angew. Chem. Int. Ed. 24 (1985) 85-93. DOI:10.1002/anie.198500851 |
| [59] |
V. Schellenberger, H.D. Jakubke, Angew. Chem. Int. Ed. 30 (1991) 1437-1449. DOI:10.1002/(ISSN)1521-3773 |
| [60] |
H.D. Jakubke, Angew. Chem. Int. Ed. 34 (1995) 175-177. DOI:10.1002/(ISSN)1521-3773 |
| [61] |
J.H. van't Hoff, Zeitschrift für, Anorg. Chem. 18 (1898) 1-13. DOI:10.1002/zaac.18980180102 |
| [62] |
C.F.W. Becker, Nat. Chem. Biol. 14 (2017) 2-3. DOI:10.1038/nchembio.2533 |
| [63] |
T.K. Chang, D.Y. Jackson, J.P. Burnier, et al., Proc. Natl. Acad. Sci. U. S. A. 91 (1994) 12544-12548. DOI:10.1073/pnas.91.26.12544 |
| [64] |
M. Rashidian, J.K. Dozier, M.D. Distefano, Bioconjugate Chem. 24 (2013) 1277-1294. DOI:10.1021/bc400102w |
| [65] |
C.B. Rosen, M.B. Francis, Nat. Chem. Biol. 13 (2017) 697-705. DOI:10.1038/nchembio.2416 |
| [66] |
D.B. Smithrud, P.A. Benkovic, S.J. Benkovic, et al., Proc. Natl. Acad. Sci. U. S. A. 97 (2000) 1953-1958. DOI:10.1073/pnas.040534397 |
| [67] |
K. Yazawa, K. Numata, Molecules 19 (2014) 13755-13774. DOI:10.3390/molecules190913755 |
| [68] |
G.K.T. Nguyen, C.T.T. Wong, J. Biochem. Chem. Sci. 2017 (2017) 1-13. |
| [69] |
X.L. Tan, L. Xu, J. Shi, et al., Prog. Chem. 26 (2014) 1741-1751. |
| [70] |
R.A. Oliver, R. Li, C.A. Townsend, Nat. Chem. Biol. 14 (2018) 5-7. |
| [71] |
M.I. Arif, A. Toplak, W. Szymanski, et al., Adv. Synth. Catal. 356 (2014) 2197-2202. DOI:10.1002/adsc.v356.10 |
| [72] |
B. Wu, H.J. Wijma, L. Song, et al., ACS Catal. 6 (2016) 5405-5414. DOI:10.1021/acscatal.6b01062 |
| [73] |
S.K. Mazmanian, Science 285 (1999) 760-763. DOI:10.1126/science.285.5428.760 |
| [74] |
H. Ton-That, G. Liu, S.K. Mazmanian, et al., Proc. Natl. Acad. Sci. U. S. A. 96 (1999) 12424-12429. DOI:10.1073/pnas.96.22.12424 |
| [75] |
M. Ritzefeld, Chem. Eur. J. 20 (2014) 8516-8529. DOI:10.1002/chem.201402072 |
| [76] |
L. Schmohl, D. Schwarzer, Curr. Opin. Chem. Biol. 22 (2014) 122-128. DOI:10.1016/j.cbpa.2014.09.020 |
| [77] |
W. van't Hof, S. Hansenova Manaskova, E.C. Veerman, et al., Biol. Chem. 396 (2015) 283-293. |
| [78] |
J.M. Antos, M.C. Truttmann, H.L. Ploegh, Curr. Opin. Struct. Biol. 38 (2016) 111-118. DOI:10.1016/j.sbi.2016.05.021 |
| [79] |
H. Mao, S.A. Hart, A. Schink, et al., J. Am. Chem. Soc. 126 (2004) 2670-2671. DOI:10.1021/ja039915e |
| [80] |
M.W. Popp, J.M. Antos, G.M. Grotenbreg, et al., Nat. Chem. Biol. 3 (2007) 707-708. DOI:10.1038/nchembio.2007.31 |
| [81] |
G.T. Hess, J.J. Cragnolini, M.W. Popp, et al., Bioconjugate Chem. 23 (2012) 1478-1487. DOI:10.1021/bc300130z |
| [82] |
L.K. Swee, C.P. Guimaraes, S. Sehrawat, et al., Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 1428-1433. DOI:10.1073/pnas.1214994110 |
| [83] |
B.M. Paterson, K. Alt, C.M. Jeffery, et al., Angew. Chem. Int. Ed. 53 (2014) 6115-6119. DOI:10.1002/anie.201402613 |
| [84] |
T. Sakamoto, S. Sawamoto, T. Tanaka, et al., Bioconjugate Chem. 21 (2010) 2227-2233. DOI:10.1021/bc100206z |
| [85] |
R. Parthasarathy, S. Subramanian, E.T. Boder, Bioconjugate Chem. 18 (2007) 469-476. DOI:10.1021/bc060339w |
| [86] |
J.M. Antos, M.W. Popp, R. Ernst, et al., J. Biol. Chem. 284 (2009) 16028-16036. DOI:10.1074/jbc.M901752200 |
| [87] |
J.G. Bolscher, M.J. Oudhoff, K. Nazmi, et al., FASEB J. 25 (2011) 2650-2658. DOI:10.1096/fj.11-182212 |
| [88] |
T. Ito, R. Sadamoto, K. Naruchi, et al., Biochemistry 49 (2010) 2604-2614. DOI:10.1021/bi100094g |
| [89] |
C.P. Guimaraes, M.D. Witte, C.S. Theile, et al., Nat. Protoc. 8 (2013) 1787-1799. DOI:10.1038/nprot.2013.101 |
| [90] |
C.S. Theile, M.D. Witte, A.E. Blom, et al., Nat. Protoc. 8 (2013) 1800-1807. DOI:10.1038/nprot.2013.102 |
| [91] |
H. Hirakawa, S. Ishikawa, T. Nagamune, Biotechnol. J. 10 (2015) 1487-1492. DOI:10.1002/biot.201500012 |
| [92] |
Y. Yamamura, H. Hirakawa, S. Yamaguchi, et al., Chem. Commun. 47 (2011) 4742-4744. DOI:10.1039/c0cc05334a |
| [93] |
J.M. Antos, G.L. Chew, C.P. Guimaraes, et al., J. Am. Chem. Soc. 131 (2009) 10800-10801. DOI:10.1021/ja902681k |
| [94] |
D.J. Williamson, M.A. Fascione, M.E. Webb, et al., Angew. Chem. Int. Ed. 51 (2012) 9377-9380. DOI:10.1002/anie.201204538 |
| [95] |
F. Liu, E.Y. Luo, D.B. Flora, et al., J. Org. Chem. 79 (2014) 487-492. DOI:10.1021/jo4024914 |
| [96] |
Y.M. Li, Y.T. Li, M. Pan, et al., Angew. Chem. Int. Ed. 53 (2014) 2198-2202. DOI:10.1002/anie.201310010 |
| [97] |
Z.M. Wu, S.Z. Liu, X.Z. Cheng, et al., Chin. Chem. Lett. 28 (2017) 553-557. DOI:10.1016/j.cclet.2016.11.001 |
| [98] |
G.K.T. Nguyen, S. Wang, Y. Qiu, et al., Nat. Chem. Biol. 10 (2014) 732-738. DOI:10.1038/nchembio.1586 |
| [99] |
G.K.T. Nguyen, A. Kam, S. Loo, et al., J. Am. Chem. Soc. 137 (2015) 15398-15401. DOI:10.1021/jacs.5b11014 |
| [100] |
X. Hemu, Y. Qiu, G.K.T. Nguyen, et al., J. Am. Chem. Soc. 138 (2016) 6968-6971. DOI:10.1021/jacs.6b04310 |
| [101] |
G.K.T. Nguyen, X. Hemu, J.-P. Quek, et al., Angew. Chem. Int. Ed. 55 (2016) 12802-12806. DOI:10.1002/anie.201607188 |
| [102] |
Y. Cao, G.K.T. Nguyen, J.P. Tam, et al., Chem. Commun. 51 (2015) 17289-17292. DOI:10.1039/C5CC07227A |
| [103] |
X. Bi, J. Yin, G.K.T. Nguyen, et al., Angew. Chem. Int. Ed. 56 (2017) 7822-7825. DOI:10.1002/anie.201703317 |
| [104] |
G.K.T. Nguyen, Y. Cao, W. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 15694-15698. DOI:10.1002/anie.201506810 |
| [105] |
Y. Cao, G.K.T. Nguyen, S. Chuah, et al., Bioconjugate Chem. 27 (2016) 2592-2596. DOI:10.1021/acs.bioconjchem.6b00538 |
| [106] |
G.K. Nguyen, Y. Qiu, Y. Cao, et al., Nat. Protoc. 11 (2016) 1977-1988. DOI:10.1038/nprot.2016.118 |
| [107] |
E.W. Schmidt, J.T. Nelson, D.A. Rasko, et al., Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 7315-7320. DOI:10.1073/pnas.0501424102 |
| [108] |
J. Lee, J. McIntosh, B.J. Hathaway, et al., J. Am. Chem. Soc. 131 (2009) 2122-2124. DOI:10.1021/ja8092168 |
| [109] |
C.J. Barber, P.T. Pujara, D.W. Reed, et al., J. Biol. Chem. 288 (2013) 12500-12510. DOI:10.1074/jbc.M112.437947 |
| [110] |
H. Luo, S.Y. Hong, R.M. Sgambelluri, et al., Chem. Biol. 21 (2014) 1610-1617. DOI:10.1016/j.chembiol.2014.10.015 |
| [111] |
R. Yang, Y.H. Wong, G.K.T. Nguyen, et al., J. Am. Chem. Soc. 139 (2017) 5351-5358. DOI:10.1021/jacs.6b12637 |
| [112] |
L. Abrahmsen, J. Tom, J. Burnier, et al., Biochemistry 30 (1991) 4151-4159. DOI:10.1021/bi00231a007 |
| [113] |
A. Toplak, T. Nuijens, P.J.L.M. Quaedflieg, et al., Adv. Synth. Catal. 358 (2016) 2140-2147. DOI:10.1002/adsc.v358.13 |
| [114] |
K. Piotukh, B. Geltinger, N. Heinrich, et al., J. Am. Chem. Soc. 133 (2011) 17536-17539. DOI:10.1021/ja205630g |
| [115] |
T. Biswas, V.S. Pawale, D. Choudhury, et al., Biochemistry 53 (2014) 2515-2524. DOI:10.1021/bi4016023 |
| [116] |
K.D. Nikghalb, N.M. Horvath, J.L. Prelesnik, et al., Chembiochem 19 (2018) 185-195. DOI:10.1002/cbic.201700517 |
| [117] |
I. Chen, B.M. Dorr, D.R. Liu, Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 11399-11404. DOI:10.1073/pnas.1101046108 |
| [118] |
H. Hirakawa, S. Ishikawa, T. Nagamune, Biotechnol. Bioeng. 109 (2012) 2955-2961. DOI:10.1002/bit.v109.12 |
| [119] |
P. Gianella, E.L. Snapp, M. Levy, Biotechnol. Bioeng. 113 (2016) 1647-1657. DOI:10.1002/bit.v113.8 |
| [120] |
K.S. Harris, T. Durek, Q. Kaas, et al., Nat. Commun. 6 (2015) 10199. DOI:10.1038/ncomms10199 |
| [121] |
L.B. Evnin, C.S. Craik, Ann. N.Y. Acad. Sci. 542 (1988) 61-74. DOI:10.1111/nyas.1988.542.issue-1 |
| [122] |
S. Liebscher, M. Schopfel, T. Aumuller, et al., Angew. Chem. Int. Ed. 53 (2014) 3024-3028. DOI:10.1002/anie.v53.11 |
| [123] |
C. Meyer, S. Liebscher, F. Bordusa, Bioconjugate Chem. 27 (2016) 47-53. DOI:10.1021/acs.bioconjchem.5b00618 |
| [124] |
S. Liebscher, P. Kornberger, G. Fink, et al., Chembiochem. 15 (2014) 1096-1100. DOI:10.1002/cbic.201400059 |
| [125] |
I. Svendsen, Carlsberg. Res. Commun. 41 (1976) 237-291. DOI:10.1007/BF02906260 |
| [126] |
C.-F. Liu, J.P. Tam, Org. Lett. 3 (2001) 4157-4159. DOI:10.1021/ol0167614 |
| [127] |
T. Nakatsuka, T. Sasaki, E.T. Kaiser, J. Am. Chem. Soc. 109 (1987) 3808-3810. DOI:10.1021/ja00246a064 |
| [128] |
D. Jackson, J. Burnier, C. Quan, et al., Science 266 (1994) 243-247. DOI:10.1126/science.7939659 |
| [129] |
S.H. Henager, N. Chu, Z. Chen, et al., Nat. Methods 13 (2016) 925-927. DOI:10.1038/nmeth.4004 |
| [130] |
A. Toplak, T. Nuijens, P.J.L.M. Quaedflieg, et al., Adv. Synth. Catal. 358 (2016) 2140-2147. DOI:10.1002/adsc.v358.13 |
| [131] |
C. Tuthill, Ann. N.Y. Acad. Sci. 1112 (2007) 351-356. DOI:10.1196/annals.1415.007 |
| [132] |
J. Li, C.H. Liu, F.S. Wang, Peptides 31 (2010) 2151-2158. DOI:10.1016/j.peptides.2010.07.026 |
| [133] |
M. Schmidt, A. Toplak, H.J. Rozeboom, et al., Org. Biomol. Chem. 16 (2018) 609-618. DOI:10.1039/C7OB02812A |
| [134] |
A.M. Weeks, J.A. Wells, Nat. Chem. Biol. 14 (2018) 50-57. |
| [135] |
C.F.W. Becker, Nat. Chem. Biol. 14 (2018) 2-3. |