 2018, Vol. 29
2018, Vol. 29 



Molecules with chroman-4-one scaffold are not only ubiquitous in natural products, but also essential nutrients for the human body [1]. Moreover, they are also widely applied in pharmaceuticals, pigments and dyes [2]. Thus, the studies of these compounds' syntheses [3] and modifications have attracted more and more attentions. A variety of bioactive molecules, which are derived from chroman-4-one, have been well applied in drugs (Fig. 1), such as the new SIRT2 inhibitor [4], flavonones [5], and GPR119 [6]. It is worthy to mention that flavonones can effectively prevent cell degeneration and delay aging process and GPR119 shows agonist activity.

|
Download:
|
| Fig. 1. Pharmaceuticals derived from chroman-4-ones | |
Moreover, the studies of fluorine-contained compounds' syntheses have become a field of great interest and great progress has been made in fluorination reactions [7] among which Selectfluor (1-chloromethyl-4-fluoro-1, 4-diazoniabicyclo[2.2.2] octane bis(tetrafluoroborate)) is the most frequently used fluorination reagents. Reports about electrophilic and free radical fluorinations, which employed Selectfluor as fluorine sources, appeared frequently [8]. In 1996, Bolós's group reported the synthesis of 3-fluoro-4H-chromen-4-one from 1-fluoro-2, 4, 6- trimethylpyridinium triflate (NFTP), which was an expensive fluorine reagent (Scheme 1a) [9]. And in 2006, Shreeve's group obtained 1, 1-difluorinated carbonyl compounds from enamines and Selectfluor (Scheme 1b) [10]. Recently, synthesis of chromone and its derivatives from o-hydroxyarylenaminones had attracted considerable attention because of their simple operation and easily accessible starting materials [11]. During this manuscript preparation, Yang and coworkers reported a base promoted cyclization of o-hydroxyarylenaminones with Selectfluor to access 3, 3- difluorochroman-4-ones [12]. Compared with their work, there are several advantages of ours: 1) without the additional of base, 2) more wider substrate scope including disubstituted o-hydroxyarylenaminone which was not accessed in yang's report, 3) lower Selectfluor loading. Therefore, we would like to report our progress on the synthesis of difluorinated chromanones with Selectfluor as a fluorine source (Scheme 1c).

|
Download:
|
| Scheme 1. Reactions of fluorine reagents with arylenaminones | |
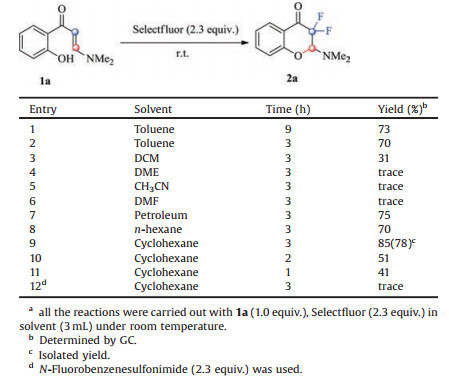
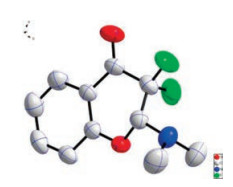
Our initial studies were carried out with o-hydroxyarylenaminone 1a and Selectfluor, and the mixture was stirred in toluene at room temperature for about 9 h. To our delight, the desired product 2-(dimethylamino)-3, 3-difluorochroman-4-one (2a) was obtained in 73% yield (Table 1, entry 1). The structure of 2a was confirmed by X-ray analysis (Fig. 2) (see Supporting information for details). When the reaction time was shortened to 3 h, the yield was almost kept untouched. To further improve the yield of 2a, various solvents were tested (Table 1, entries 3-8). It turned out that solvent affected the reaction greatly, and non-polar solvents afforded much higher yields than polar ones. Polar solvents such as DME, CH3CH, DMF could barely afforded any product (Table 1, entries 4-6), however, non-polar solvents such as petroleum, hexane could gave 2a in acceptable yields (Table 1, entries 7 and 8). To our delight, when cyclohexane was used as solvent, 2a could be obtained in 85% yield (Table 1, entry 9). When the reaction time was further shortened, the yield of 2a decreased significantly (Table 1, entries 10 and 11). Other fluorine reagent, like NFSI, was also tested in our reaction systems, unfortunately no desired product was detected (Table 1, entry 12). By far, the optimal reaction conditions were obtained as: 1a (1.0 equiv.) and Selectfluor (2.3 equiv.) were stirred in cyclohexane (3 mL) at room temperature for 3 h. And, typical experimental procedure was below: To a 25 mL Schlenk tube, under a nitrogen atmosphere, was added 0.20 mmol of o-hydroxyarylenaminone (1a) and 0.46 mmol of Selectfluor (2.3 equiv.), then cyclohexane (3 mL) was added by syringe. The reaction mixture was stirred violently at room temperature until yellow emulsion to become clear, even colorless (3-4 h generally needed). Then, the reaction mixture was filtered by the sand core funnel. Subsequently, the filtrate was concentrated to dryness and purified by silica gel chromatography (silica gel, PE: Et3N = 50:1, v/v) to afford the product 2a (36 mg, 78%) as a white solid.

|
Download:
|
| Fig. 2. The X-ray crystallography of 2a | |
|
|
Table 1 Optimization of the reaction conditions.a |
{kind=link}
{kind=link}
{kind=link}
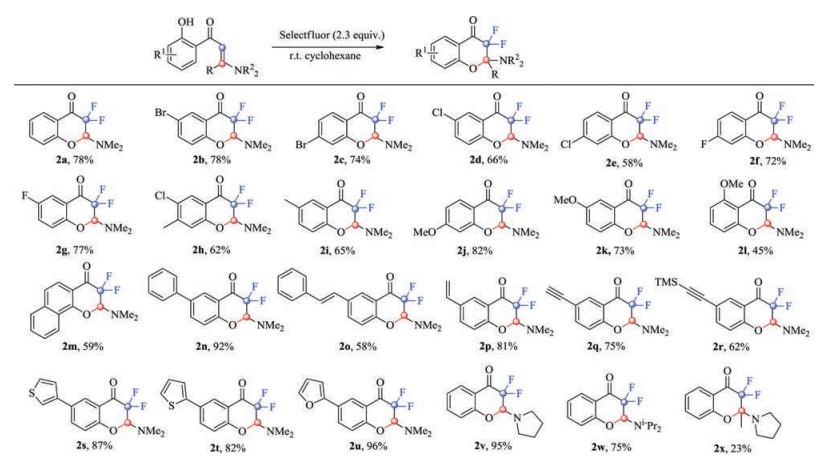
With the optimized conditions in hand, the scope of the substrate was studied (Scheme 2). Substituents on the phenyl rings had little effect on the yields of products, either electronwithdrawing or electron-donating groups could go through the reactions smoothly, and afforded the desired products in moderate to high yields (2a-2k). However, the position of some substituents on the phenyl rings has a great impact on the reaction (2j-2l) with the ortho one giving the lowest yield, and the steric hindrance might be the direct cause of the low yield of 2l. Polycyclic substrates could also go through the cyclization and difluorination process smoothly rendering 2m in 59% yield, and 2n in 92% yield. Interestingly, when vinyl and alkynyl groups were placed on the phenyl rings of the substrates, the desired products were also successfully obtained (2o-2r). It is worthy to note that both terminal and internal unsaturated carbon-carbon bonds were well compatible. Heterocyclic substituents, such as 3-thienyl, 2- thienyl and 2-furyl, were also tolerated well under our reaction systems, and the desired products were obtained in good to excellent yields (2s-2u). The substituents on the nitrogen atom were also tested (2v, 2w). When N, N-dimethyl group was replaced with pyrrolidinyl, the corresponding product 2v was obtained in 95% yield, which was much higher than the yield of 2a. However, when replaced with diisopropyl, the yield of 2w was decreased a little compared with 2a. At last, when large steric hindered substrate 1x was applied, the desired product 2x was only afforded in 23% yield.

|
Download:
|
| Scheme 2. The scope of the substrates | |
{kind=link}
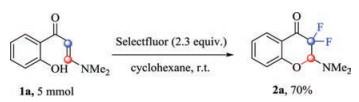
Large scale experiment was also carried out with 1a (5 mmol) and Selectfluor (11.5 mmol) under our standard reaction conditions (Scheme 3). The reaction was completed in 6 h with 70% of 2a.

|
Download:
|
| Scheme 3. Large scale experiment | |
{kind=link}
To gain a better understanding of the reaction process, several control experiments were conducted (Scheme 4). When the reaction was carried out in the presence of TMEPO (2.3 equiv.) under our standard conditions, difluorinated product 2a could still be isolated in 45% yield (Scheme 4a). Thus, we considered that the reaction could not proceed via single electron transfer (SET) radical reaction mechanism. Next, substrates 1y and 1z were investigated under the standard reaction conditions, and no desired products were obtained, which illustrated the importance of amino groups (Schemes 4b and c). So we deem that the reaction might go through via iminium intermediate (Scheme 5). Meantime, it could also explain why product 2x was only obtained in 23% yield, since the presence of methyl group might hinder the formation of iminium intermediate at a great extent [10]. At last, 3a was synthesized and exposed to the standard conditions, however, no difluorinated product was detected (Scheme 4d). The result illustrated that the difluorination step went before the cyclization process (Scheme 5).

|
Download:
|
| Scheme 4. Control experiments | |
{kind=link}

|
Download:
|
| Scheme 5. Plausible mechanism | |
{kind=link}
A proposed mechanism for the synthesis of 3, 3-difluorochroman-4-ones was shown in Scheme 5. It is deemed that species A might exist in our reaction system, which could swiftly react with the electrophilic Selectfluor, affording species B. Then, HBF4 is released and affords intermediate C. Due to the electron withdrawing character of fluorine, species D is easier to be generated, which reacted with Selectfluor again and affords species E. Intramolecular cyclization of E affords product 2a along with the release of molecular HBF4.
In conclusion, a facile and gentle strategy for the synthesis of 3, 3-difluorochroman-4-ones from o-hydroxyarylenaminones and Selectfluor was developed in our laboratory. The reaction processed swiftly in cyclohexane at room temperature under catalyst-free conditions and completed in 3 h. More importantly, the products were obtained in moderate to high yields with excellent chemselectivity. Various kinds of functional groups were tolerated, especially vinyl and alkynyl groups, which were kept untouched under standard conditions. The obtained difluorinated products were useful and their applications were under studies in our laboratory.
AcknowledgmentsFinancial support from the Recruitment Program of Global Experts (1000 Talents Plan), the Natural Science Foundation of Fujian Province (No. 2016J01064), Fujian Hundred Talents Plan, Program of Innovative Research Team of Huaqiao University (No. Z14X0047) and Subsidized Project for Cultivating Postgraduates' Innovative Ability in Scientific Research of Huaqiao University (for Z. Kuang) are gratefully acknowledged. We also thank the Instrumental Analysis Center of Huaqiao University for analysis support.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2017.10.027.
| [1] |
(a) D. G. Wei, G. F. Yang, J. Wan, C. G. Zhan, J. Agr. Food Chem. 53(2005) 1604-1611; (b) L. Feng, M. M. Maddox, M. Zahidul Alam, et al., J. Med. Chem. 57(2014) 8398-8420. |
| [2] |
A. Gaspar, M. João Matos, J. Garrido, E. Uriarte, F.B. Chromone, Eur. J. Med. Chem. 78 (2014) 340-374. DOI:10.1016/j.ejmech.2014.03.047 |
| [3] |
(a) M. M. Biddle, M. Lin, K. A. Scheidt, J. Am. Chem. Soc. 129(2007) 3830-3831; (b) Z. Zhang, C. Pan, Z. Wang, Chem. Commun. 43(2007) 4686-4688; (c) L. Meng, M. Y. Jin, J. Wang, Org. Lett. 18(2016) 4986-4989; (d) K. Tanaka, T. Sugino, Green Chem. 3(2001) 133-134. |
| [4] |
Fride'n-Saxin M., T. Seifert, M.R. Landergren, et al., J. Med. Chem. 55 (2012) 7104-7113. DOI:10.1021/jm3005288 |
| [5] |
debielle M.M., R. Keirouz, E. Okada, D. Shibata, W.R. Dolbier, Tetrahedron Lett. 49 (2008) 589-593. DOI:10.1016/j.tetlet.2007.11.146 |
| [6] |
T. Ogawa, S., Tanaka, A. Okano, et al., WO 2012077655 A1, 2012.
|
| [7] |
(a) C. Ni, M. Hu, J. Hu, Chem. Rev. 115(2015) 765-825; (b) X. H. Xu, K. Matsuzaki, N. Shibata, Chem. Rev. 115(2015) 731-764; (c) J. Charpentier, N. Früh, A. Togni, Chem. Rev. 115(2015) 650-682. |
| [8] |
(a) R. T. Thornbury, V. Saini, T. A. Fernandes, et al., Chem. Sci. 8(2017) 2890-2897; (b) T. J. Barker, D. L. Boger, J. Am. Chem. Soc. 134(2012) 13588-13591; (c) C. Zhang, Z. Li, L. Zhu, et al., J. Am. Chem. Soc. 135(2013) 14082-14085; (d) L. Zhu, H. Chen, Z. Wang, C. Li, Org. Chem. Front. 1(2014) 1299-1305; (e) X. H. Yu, X. H. Xu, F. L. Qing, Adv. Synth. Catal. 357(2015) 2039-2044; (f) Y. Li, X. Jiang, C. Zhao, et al., ACS Catal. 7(2017) 1606-1609; (g) H. Wang, L. N. Guo, X. H. Duan, Chem. Commun. 50(2014) 7382-7384. |
| [9] |
J. Bolós, S. Gubert, L. Anglada, et al., J. Med. Chem. 39 (1996) 2962-2970. DOI:10.1021/jm950894b |
| [10] |
W. Peng, J. M. Shreeve, J. Org. Chem. 70(2005) 5760-5763.
|
| [11] |
(a) H. Xiang, C. Yang, Org. Lett. 16(2014) 5686-5689; (b) H. Xiang, Q. Zhao, Z. Tang, et al., Org. Lett. 19(2017) 146-149; (c) M. O. Akram, S. Bera, N. T. Patil, Chem. Commun. 52(2016) 12306-12309; (d) X. Z. Zhang, D. L. Ge, S. Y. Chen, X. Q. Yu, RSC Adv. 6(2016) 66320-66323; (e) S. Zhong, Y. Liu, X. Cao, J. P. Wan, ChemCatChem 9(2017) 465-468; (f) S. Wan, Y. Zhong, Eur. J. Org. Chem. 2017(2017) 4401-4404. |
| [12] |
Q. Zhao, H. Xiang, J.A. Xiao, et al., J. Org. Chem. 82 (2017) 9837-9843. DOI:10.1021/acs.joc.7b01339 |