 2018, Vol. 29
2018, Vol. 29 



b Department of Chemistry, Shantou University, Shantou 515063, China
Iron-based catalysts have drawn much attention because they are earth abundant, cheap, and environment benign [1]. In recent years, iron-based complexes have been explored for a variety of transformations, such as hydrogenation [1, 2], hydrosilylation [3-5], hydroboration [6-8], proton reduction [9], H2 splitting [10-12], and CO2 reduction [13]. Significantly, iron hydride species was thought to be the key intermediate in those transformations [14].
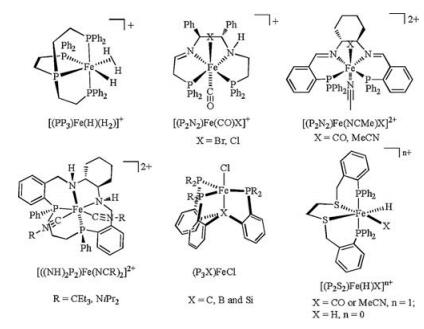
Iron complexes with tetradentate ligand have exhibited high reactivity for both stoichiometric and catalytic reactions [15-22]. For example, iron-based complexes with different types of diimino diphosphine ligands, as known as P2N2, have shown high reactivity in asymmetric hydrogenation and asymmetric transfer hydrogenation of ketones and imines [15, 16]. The P2N2 type ligand is usually coordinated to the Fe center in a planar arrangement which results in two active sites trans to one another in an octahedral geometry (Fig. 1). Unlike P2N2, diphosphinodithio ligand (P2S2) was found to coordinate to iron center in a folded way that the two active sites are in a cis-α disposition. Although the reactivity of iron (Ⅱ) hydrides in [(P2S2)Fe(H)X]n+ series (X = CO, MeCN or H-) has been investigated [23], they are inactive in catalytic hydroboration or hydrosilylation of organic carbonyl substrates.

|
Download:
|
| Fig. 1. Structures of iron(Ⅱ) complexes with tetradentate ligands. | |
By switching one of S atoms in P2S2 to N, we describe here a series of well-characterized iron(Ⅱ) hydride complexes supported by PSNP tetradentate ligands. The combination of soft-hard donating atoms stabilizes such iron(Ⅱ) hydrides for investigations and increases their reactivity for catalysis, i.e., catalytic hydroboration of aldehydes at mild conditions.
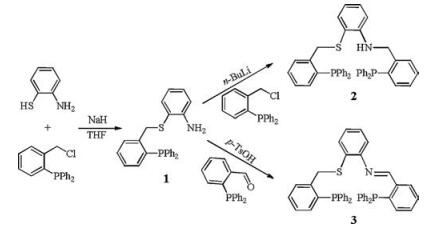
The tetradentate ligands 2 and 3 were synthesized from the same precursor, compound 1 (Scheme 1). In the presence of NaH, the reaction of 1, 2-NH2C6H4SH with 2-(diphenylphosphino)benzyl chloride [24, 25] afforded 1 in 95% yield. Treatment of 1 with 1.1 equiv. of n-butyllithium and further reaction with 2-(diphenylphosphino)benzyl chloride produced 2 (yield: 70%). Condensation of 1 and 2-(diphenylphosphino)benzaldehyde in the presence of 10% p-toluenesulfonic acid gave ligand 3. The 31P NMR spectrum of 1 exhibits a single peak at δ -17.0 (Fig. S2 in Supporting information), while NH2 protons are shown as a broad peak at d 4.08 in 1H NMR spectrum (Fig. S1 in Supporting information). The 31P NMR spectrum of 2 displayed two phosphorus resonances at δ -15.9 and -17.2 (Fig. S4 in Supporting information). By contrast, the 31P signals of 3 were observed at δ -14.4 and -16.7 (Fig. S6 in Supporting information).

|
Download:
|
| Scheme 1. Synthetic route of the ligands 2 and 3. | |
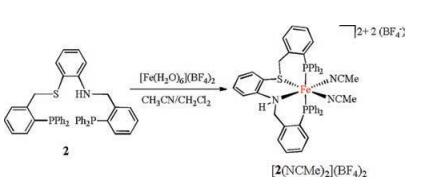
Treatment of the CH2Cl2/MeCN (1:1, v/v) solution of 2 with 1 equiv. of [Fe(H2O)6](BF4)2 provided bis(acetonitrile) iron complex [2(NCMe)2](BF4)2, which was isolated as pink solids in 86% yield (Scheme 2). The 31P NMR spectrum shows two doublets peak at δ 37.4 and 29.6 (JP-P = 121.3 Hz) (Fig. S8 in Supporting information). Such a large 31P-31P coupling constant suggests the two P atoms are trans to each other.

|
Download:
|
| Scheme 2. Synthesis of bis(acetonitrile) iron complex [2(NCMe)2](BF4)2. | |
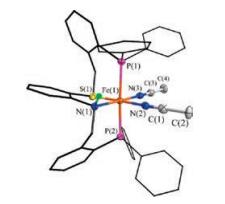
By diffusion of diethyl ether into a MeCN solution of [2(NCMe)2] (BF4)2, single crystals suitable for X-ray diffraction were obtained (Fig. 2). Crystallographic analysis [2(NCMe)2](BF4)2 reveals that the iron center is coordinated to the folded PSNP ligand and two MeCN molecules, which are in cis position. In the pseudo-octahedral geometry, the S and N atoms in PSNP and another two N atoms in MeCN groups are essentially coplanar. Two P atoms are trans to the plane with a P-Fe-P angle of 176.42(5)°, which is in agreement with the observations in the 31P NMR spectrum. The structure of [2(NCMe)2](BF4)2 is very similar to [Fe(P2S2)(NCMe)2](CF3SO3)2 and Fe(P2O2)(CO)2 complexes described previously [3, 23, 26].

|
Download:
|
| Fig. 2. Structure (50% thermal ellipsoids) of [2(NCMe)2](BF4)2. For clarity, the anion and hydrogen atoms bonded to carbon atoms are omitted. Selected distances (Å) and angles (deg.): Fe(1)-P(1) 2.3398(1), Fe(1)-P(2) 2.3062(1), Fe(1)-S(1) 2.2276(1), Fe(1)-N(1) 2.059(3), Fe(1)-N(2) 1.930(4), Fe(1)-N(3) 1.915(3), N(2)-C(1) 1.135(5), N (3)-C(3) 1.136(5), P(1)-Fe(1)-P(2) 176.42(5), N(1)-Fe(1)-S(1) 86.69(1), N(2)-Fe(1)-N (3) 91.05(1). | |
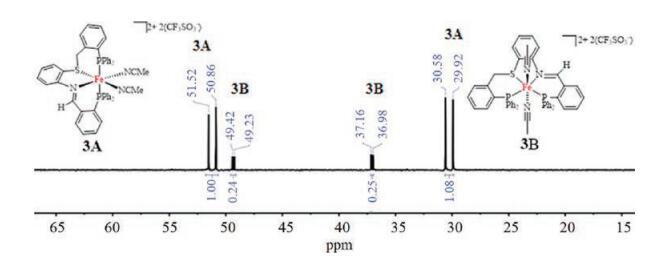
Complex [3(NCMe)2](CF3SO3)2 was prepared by the reaction of 3 with Fe(CF3SO3)2 in MeCN/CH2Cl2 (1:1, v/v) at room temperature. The 31P NMR spectrum of [3(NCMe)2](CF3SO3)2 exhibits four sets of doublets, which are assigned as two isomers 3A and 3B (Fig. 3). The ratio of 3A/3B is approximately 4:1. The two doublets at δ 51.2 and 30.3 with JP-P = 132.7 Hz is indicative of two unique trans P atoms in 3A [3, 26]. The 31P signals for 3B at δ 49.3 and 37.1 with much smaller 31P-31P coupling constant JP-P = 35.7 Hz compared to that for 3A. In 3B, the PSNP ligand is probably coordinated to the Fe center in a planar arrangement, which is commonly found for [Fe(P2N2)(NCMe)2]2+ cationic complex [27]. These results suggest the PSNP ligand is flexible to achieve different coordination modes.

|
Download:
|
| Fig. 3. 31P NMR spectrum of [3(NCMe)2](CF3SO3)2 exhibiting two isomers 3A and 3B. | |
The structure of 3A was confirmed by X-ray crystallography (Fig. 4). Similar to that of [2(NCMe)2](BF4)2, the Fe center in 3A is coordinated to a folded PSNP ligand and adopts a quasi-octahedral geometry. In 3A, the P-Fe-P angle 171.90(4)° is slightly smaller than 176.42(5)° for [2(NCMe)2](BF4)2. Upon dissolving the crystals of 3A in CD3CN at room temperature, the 31P{1H} NMR spectrum displayed the phosphorus resonances for both 3A and 3B in the ratio of 4:1 that is the same obtained from the synthesis. The ratio of 3A and 3B does not change over the time of 24 h. However, the attempts to obtain the structure of 3B by single crystal X-ray diffraction were unsuccessful.

|
Download:
|
| Fig. 4. Structure (50% thermal ellipsoids) of 3A. For clarity, the anion and hydrogen atoms are omitted. Selected distances (Å) and angles (deg.): Fe(1)-P(1) 2.3448(9), Fe (1)-P(2) 2.2803(9), Fe(1)-S(1) 2.2332(9), Fe(1)-N(1) 1.986(3), Fe(1)-N(2) 1.934(3), Fe (1)-N(3) 1.915(3), N(2)-C(1) 1.132(4), N(3)-C(3) 1.135(4) P(1)-Fe(1)-P(2) 171.90(4), N (1)-Fe(1)-S(1) 85.43(8), N(2)-Fe(1)-N(3) 91.05(1). | |
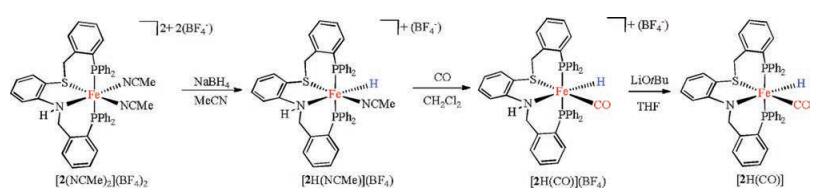
The iron hydrides were synthesized and investigated based on [2(NCMe)2](BF4)2, which exhibit no isomers. Treatment of [2(NCMe)2](BF4)2 with one equiv. of NaBH4 gave cationic iron(Ⅱ) hydride [2H(NCMe)](BF4) (Scheme 3). In the 1H NMR spectrum, the hydride ligand displays its characteristic triplet signal at δ -19.4 (JP-H = 63.2 Hz) (Fig. S13 in Supporting information). From IR spectrum, the νFe-H band was observed at 1823 cm-1 that is consistent with 1849 cm-1 for HFe(dppe)2Cl [26]. The 31P NMR spectrum of hydrido acetonitrile complex displayed four sets of doublets peaks (δ 64.3, 63.7 (d, JP-P = 52.5 Hz), and δ 57.1, 56.5 (δ, JP-P = 54.5 Hz)) (Fig. S14 in Supporting information).

|
Download:
|
| Scheme 3. Syntheses of iron(Ⅱ) hydrides. | |
The solid state structure of [2H(NCMe)](BF4) was confirmed by X-ray diffraction (Fig. 5). The six-coordinate iron resides in a distorted octahedral geometry reflected by its P-Fe-P angle of 153.27(6)°. The hydride ligand was located in the difference map and its position and isotropic displacement parameters were refined. In the structure, the hydride ligand is trans to the amine group. The Fe-H bond length is 1.46(5) Å, consistent with those observed for low-spin iron(Ⅱ) hydrides [5, 23, 28].

|
Download:
|
| Fig. 5. Structure (50% thermal ellipsoids) of [2H(NCMe)](BF4). For clarity, the anion and hydrogen atoms bonded to carbon atoms are omitted. Selected distances (Å) and angles (deg.): Fe(1)-H 1.46(5), Fe(1)-P(1) 2.2104(2), Fe(1)-P(2) 2.2031(2), Fe(1)- S(1) 2.2442(2), Fe(1)-N(1) 2.097(4), Fe(1)-N(2) 1.908(5), N(2)-C(1) 1.146(6), P(1)-Fe (1)-P(2) 153.27(6), N(1)-Fe(1)-S(1) 83.29(1), N(2)-Fe(1)-H 90.3(2). | |
The CH3CN ligand in [2H(NCMe)](BF4) can be replaced by CO to form hydrido carbonyl iron complex [2H(CO)](BF4). Exposure of a solution of [2H(NCMe)](BF4) to CO gas resulted in the color of the solution changed from orange to yellow over the course of 3 h. A strong nCO band was observed at 1953 cm-1 in IR spectrum (Fig. S19 in Supporting information). The 1H NMR spectrum displayed two hydride resonances at δ -11.5 (t, JP-H = 55.0 Hz) and -18.0 (t, JP-H = 52.4 Hz, Fig. S16 in Supporting information), which indicate that two isomers exist in solution. One of the isomers of [2H(CO)](BF4) was also crystallographically characterized (Fig. 6). The substitution of MeCN ligand by CO elongates the average Fe-P distance by 0.02 Å.

|
Download:
|
| Fig. 6. Structure (50% thermal ellipsoids) of [2H(CO)](BF4). For clarity, the anion and hydrogen atoms bonded to carbon atoms are omitted. Selected distances (Å) and angles (deg.): Fe(1)-H 1.50(6), Fe(1)-P(1) 2.2337(2), Fe(1)-P(2) 2.2240(2), Fe(1)-S(1) 2.244(2), Fe(1)-N(1) 2.094(6), Fe(1)-C(1) 1.737(7), O(1)-C(1) 1.174(9), P(1)-Fe(1)-P (2) 170.20(7) N(1)-Fe(1)-S(1) 86.60(2), C(1)-Fe(1)-H 93(2). | |
Interestingly, the NH proton in [2H(CO)](BF4) undergoes deprotonation in the presence of the base, giving a neutral iron(Ⅱ) hydride [2H(CO)]. Treatment of the THF solution of [2H(CO)](BF4) with LiOtBu led the color of the solution to change immediately from yellow to dark red. The reaction process was monitored by IR spectra. The νCO band for [2H(CO)] was displayed at 1917 cm-1 (Fig. S25 in Supporting information), 36 cm-1 higher energy shift compared to that of the cationic precursor [2H(CO)](BF4). The neutral compound [2H(CO)] was characterized crystallographically (Fig. 7). In [2H(CO)], the Fe(1)-N(1) distance 2.003(6) Å is shorter by 0.091 Å in contrast to [2H(CO)](BF4). The Fe-H bond distance 1.62(4) Å in [2H(CO)] is longer than that in [2H(CO)]+ by 0.12 Å.

|
Download:
|
| Fig. 7. Structure (50% thermal ellipsoids) of [2H(CO)]. For clarity, the hydrogen atoms bonded to carbon atoms are omitted. Selected distances (Å) and angles (deg.): Fe(1)-H 1.62(4), Fe(1)-P(1) 2.2281(2), Fe(1)-P(2) 2.2104(2), Fe(1)-S(1) 2.2515 (2), Fe(1)-N(1) 2.003(6), Fe(1)-C(1) 1.721(7), O(1)-C(1) 1.178(7), P(1)-Fe(1)-P(2) 172.18(8), N(1)-Fe(1)-S(1) 85.17(2), C(1)-Fe(1)-H 89.2(2). | |
Organic boronate esters are common borylating agents applied in Suzuki-Miyaura reactions [29]. Significant efforts have been made for developing highly efficient methods to synthesize organoborane compounds based on cheap-metal catalysts [30-33]. We found [2H(NCMe)](BF4) can serve as a highly efficient catalyst for aldehydes hydroboration by pinacolborane (HBpin). At a loading of 1 mol% in benzene, [2H(NCMe)] (BF4) catalyzed benzaldehyde hydroboration in up to quantitative conversion within 20 min. In the absence of [2H(NCMe)](BF4), a trace of the hydroborated products was observed (< 5%) at room temperature in 30 min, judging from 1H NMR spectrum.
The scope of aldehyde substrates was further investigated, as summarized in Table 1. Benzaldehyde derivatives with electronwithdrawing or electron-donating groups at para- or ortho-position in the aromatic ring (entries 1-6) convert to the corresponding borate esters almost quantitatively. Not limited to aromatic aldehydes, aliphatic aldehydes were also reduced by HBpin to the borate esters quantitatively (entries 7 and 8). Functional groups such as alkenyl (entries 9-11), ferrocenyl (entry 12), pyridyl (entry 13) are all compatible with this catalytic hydroboration. For ketones, there was no hydroboration product observed for acetophenone or benzophenone.
|
|
Table 1 Hydroboration of aldehydes catalyzed by hydrido iron complexes. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
We found that [2(NCMe)2](BF4)2, [2H(CO)](BF4), and [2H(CO)] are inactive toward catalytic aldehydes hydroboration. The insertion of carbonyl group into M-H bond was proposed to be involved in transition metal catalyzed aldehydes hydroboration [30]. The MeCN ligand in [2H(NCMe)](BF4) is labile, which not only can be replaced by CO but also exchanges with CD3CN as evidenced by 1H NMR spectroscopic analysis (Fig. S45 in Supporting information). It is more likely that the coordinative site of MeCN is a potential site of binding the substrate for catalysis. The first step of the catalysis probably consists of the coordination of aldehyde to the iron(Ⅱ) center forming a η2-C=O π coordinated complex as a transition state [20]. Such coordination was thought to facilitate the subsequent transformations in an insertion/ s-bond metathesis type mechanism [30, 34]. The possibility of "Lewis-acid promoted" mechanism was not proved, at least there was no reaction observed for the stoichiometric reaction of [2H(NCMe)](BF4) with 4-methylbenzaldehyde (Fig. S46 in Supporting information).
In summary, we have described a new class of iron(Ⅱ) hydrides based on a PSNP tetradentate ligand. The iron(Ⅱ) complexes were fully characterized, and each features a folded PSNP ligand with cis reactive sites. Especially, [2H(NCMe)](BF4) has excellent catalytic activities for the hydroboration of aldehydes by HBpin at room temperature. A variety of aldehydes with different functional groups are compatible. Although the catalytic mechanism requires further investigation, iron catalysis based on such a folded coordination mode is possible.
AcknowledgmentsWe gratefully acknowledge financial support from the "1000 Youth Talents Plan", the National Natural Science Foundation of China (Nos. 21402107, 91427303), and the Natural Science Foundation of Shandong Province (Nos. ZR2014M011). We also thank Mr. Maofu Pan for assistance with the X-ray crystallography.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2017.09.059.
| [1] |
P.J. Chirik, Acc. Chem. Res. 48 (2015) 1687-1695. DOI:10.1021/acs.accounts.5b00134 |
| [2] |
T. Zell, D. Milstein, Acc. Chem. Res. 48 (2015) 1979-1994. DOI:10.1021/acs.accounts.5b00027 |
| [3] |
W.Y. Chu, X. Zhou, T.B. Rauchfuss, Organometallics 34 (2015) 1619-1626. DOI:10.1021/om501152h |
| [4] |
D. Peng, Y. Zhang, X. Du, et al., J. Am. Chem. Soc. 135 (2013) 19154-19166. DOI:10.1021/ja404963f |
| [5] |
P. Bhattacharya, J.A. Krause, H. Guan, Organometallics 30 (2011) 4720-4729. DOI:10.1021/om2005589 |
| [6] |
J. Chen, T. Xi, Z. Lu, Org. Lett. 16 (2014) 6452-6455. DOI:10.1021/ol503282r |
| [7] |
L. Zhang, D. Peng, X. Leng, Z. Huang, Angew. Chem. Int. Ed. 52 (2013) 3676-3680. DOI:10.1002/anie.201210347 |
| [8] |
J.V. Obligacion, P.J. Chirik, Org. Lett. 15 (2013) 2680-2683. DOI:10.1021/ol400990u |
| [9] |
S. Kaur-Ghumaan, L. Schwartz, R. Lomoth, et al., Angew. Chem. Int. Ed. 49 (2010) 8033-8036. DOI:10.1002/anie.v49:43 |
| [10] |
T. Liu, Q. Liao, M. O'Hagan, et al., Organometallics 34 (2015) 2747-2764. DOI:10.1021/om501289f |
| [11] |
J.M. Darmon, S. Raugei, T. Liu, et al., ACS Catal. 4 (2014) 1246-1260. DOI:10.1021/cs500290w |
| [12] |
T. Liu, D.L. Dubois, R.M. Bullock, Nat. Chem. 5 (2013) 228-233. DOI:10.1038/nchem.1571 |
| [13] |
C. Costentin, M. Robert, J.M. Saveant, Acc. Chem. Res. 48 (2015) 2996-3006. DOI:10.1021/acs.accounts.5b00262 |
| [14] |
H. Nakazawa, M. Itazaki, Top. Organomet. Chem. 33 (2011) 27-81. DOI:10.1007/978-3-642-14670-1 |
| [15] |
R.H. Morris, Acc. Chem. Res. 48 (2015) 1494-1502. DOI:10.1021/acs.accounts.5b00045 |
| [16] |
Y.Y. Li, S.L. Yu, W.Y. Shen, J.X. Gao, Acc. Chem. Res. 48 (2015) 2587-2598. DOI:10.1021/acs.accounts.5b00043 |
| [17] |
H. Fong, J.C. Peters, Inorg. Chem. 54 (2015) 5124-5135. |
| [18] |
R. Bigler, A. Mezzetti, Org. Lett. 16 (2014) 6460-6463. DOI:10.1021/ol503295c |
| [19] |
J.X. Gao, H.L. Wan, W.K. Wong, M.C. Tse, W.T. Wong, Polyhedron 15 (1996) 1241-1251. DOI:10.1016/0277-5387(95)00385-1 |
| [20] |
C. Bianchini, E. Farnetti, M. Graziani, M. Peruzzini, A. Polo, Organometallics 12 (1993) 3753-3761. DOI:10.1021/om00033a054 |
| [21] |
C. Zang, Y. Liu, Z.J. Xu, et al., Angew. Chem. Int. Ed. 55 (2016) 10253-10257. DOI:10.1002/anie.201603410 |
| [22] |
X. Gu, Y. Zhang, Z.J. Xu, C.M. Che, Chem. Commun. 50 (2014) 7870-7873. DOI:10.1039/c4cc01631a |
| [23] |
J. Liu, F. Zhang, A. Zhang, et al., Chem. Asian J. 11 (2016) 2271-2277. DOI:10.1002/asia.v11.16 |
| [24] |
J. Herrmann, P.S. Pregosin, R. Salzmann, A. Albinati, Organometallics 14 (1995) 3311-3318. DOI:10.1021/om00007a035 |
| [25] |
T.B. Rauchfuss, F.T. Patino, D.M. Roundhill, Inorg. Chem. 14 (1975) 652-656. DOI:10.1021/ic50145a043 |
| [26] |
H. Lei, A.M. Royer, T.B. Rauchfuss, D. Gray, Organometallics 31 (2012) 6408-6414. DOI:10.1021/om300631a |
| [27] |
A.A. Mikhailine, R.H. Morris, Inorg. Chem. 49 (2010) 11039-11044. DOI:10.1021/ic101548j |
| [28] |
M. Aresta, P. Giannoccaro, M. Rossi, A. Sacco, Inorg. Chim. Acta 5 (1971) 115-118. DOI:10.1016/S0020-1693(00)95893-6 |
| [29] |
A. Suzuki, Angew. Chem. Int. Ed. 50 (2011) 6722-6737. DOI:10.1002/anie.201101379 |
| [30] |
S. Chakraborty, H. Guan, Dalton Trans. 39 (2010) 7427-7436. DOI:10.1039/c002942d |
| [31] |
C. Wang, C. Wu, S. Ge, ACS Catal. 6 (2016) 7585-7589. DOI:10.1021/acscatal.6b02654 |
| [32] |
W. Yao, H. Fang, S. Peng, et al., Organometallics 35 (2016) 1559-1564. DOI:10.1021/acs.organomet.6b00161 |
| [33] |
X. Chen, Z. Lu, Org. Biomol. Chem. 15 (2017) 2280-2306. DOI:10.1039/C6OB02817A |
| [34] |
S. Chakraborty, J.A. Krause, H. Guan, Organometallics 28 (2009) 582-586. DOI:10.1021/om800948f |