 2018, Vol. 29
2018, Vol. 29 



The scope and stereoselectivities achieved by organocatalysts had grown remarkably over the past decade. Organocatalytic reactions are usually considered as operationally easy and ecofriendly because the use of metals is avoided. Organocatalytic enantioselective systems have rapidly grown and found to be a very exciting field in organic chemistry and chiral secondary amines are perhaps the most frequently used organocatalysts, which activate substrates either by raising the highest occupied molecular orbital energy level or lowering the lowest unoccupied molecular orbital energy level [1].
Heterocycles plays a vital role in the design and discovery of new physiologically active compounds [2]. The benzopyrans also belong among these privileged structures, as depict and revealed by Nicolaou et al. [3, 4]. The condensation reactions between Michael acceptors and salicylaldehydes have confirmed to be a useful route to benzopyrans [5]. Although asymmetric methods would furnish enantiomerically enriched chromenes, their development has proven to be a synthetic challenging task. By taking advantage of the capability of chiral proline derivatives to participate in the reversible formation of enamine and iminium intermediates, Enders Yamamoto, Jorgenson and List have independently developed novel types of organocatalyzed cascade reactions [1, 6, 7]. Michael addition initiated cascade Michael-aldol processes serve as powerful methods for the generation of complex structures.
Compared with the proline amide catalyst, proline with thiourea moiety having pyrrolidine ring, two hydrogen atoms on thiourea involved in hydrogen bonding and aromatic or cyclic amine as general base are functional centers in the backbone of the catalyst. It exerts stronger influence on the orientation of the iminium intermediate formed between α, β-unsaturated carbonyl and organocatalyst to enhance the stereoselectivities for the domino oxa-Michael-aldol reaction.
Herein we reported the asymmetric synthesis of chiral chromenes via organocatalytic domino oxa-Michael-aldol reaction using proline based chiral organocatalyst. The strategy we presented here is the utilization of a proline based chiral organocatalyst as a promoter for activation of the Michael acceptor 5 in a highly enantioselective controlled manner (Table 1). As our earlier interest in synthesis and study of proline based chiral organocatalysts [8], we envisioned that the iminium interaction between chiral amino catalysts and α, β-unsaturated carbonyl group was beneficial along with thiourea group as hydrogen bond donor, heterocyclic amines as general base in the domino oxaMichael-aldol reaction of simple α, β-unsaturated aromatic aldehydes and salicylaldehydes.
|
|
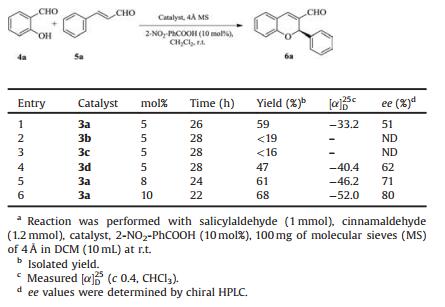
Table 1 Screening of the catalyst and catalytic loading for the domino oxa-Michael-aldol reactiona. |
All solvents were used as commercial anhydrous grade without further purification. Aluminium sheets 20 cm × 20 cm, Silica gel 60 F254, Merck grade was used for thin layer chromatography to determine the progress of reaction. The column chromatography was carried out over silica gel (80–120 mesh). Optical rotations were measured on a Polax-2L digital polarimeter. 1H NMR and 13C NMR spectra were recorded on a Bruker 300 MHz spectrometer.Melting points were measured in open capillary. Enantiomeric purity is determined on PerkinElmer Series 200 HPLC systems with chiral HPLC.
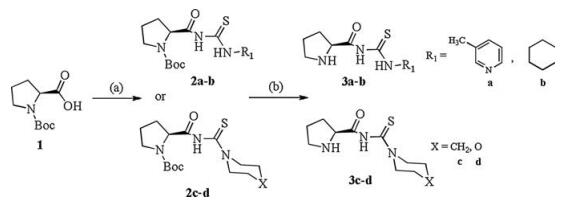
The procedures for preparing proline based chiral organocatalysts 3a–d were outlined in Scheme 1. First, commercially available Boc-L-proline reacted with thionyl chloride in DCE solvent and then the solvent was evaporated to remove excess of thionyl chloride and solvent to obtain chloride of Boc-L-proline. It was then treated with the ammonium isothiocyanate followed by addition of certain amount of the amine to obtain product. After, the N-Boc protecting group was removed to get proline based chiral organocatalysts 3a–d (detail information for preparation of 3a–d see Supporting information).

|
Download:
|
| Scheme 1. Reagent and conditions: (a) Thionyl chloride, DCE, NH4SCN, PEG-400, tosyl chloride, Et3N, R-NH2, r.t., 24 h; (b) TFA, DCM, 0 ℃. | |
Preliminary studies were carried out on the model reaction between trans-cinnamaldehyde and salicylaldehyde. The Screening of the catalyst and catalytic loading on the yield and enantioselectivity of the domino oxa-Michael-aldol reaction between salicylaldehyde and trans-cinnamaldehyde studied and results were summarized in Table 1. The organocatalysts 3b and 3c were found to be poor catalysts for this reaction because of less product formation and poor enantioselectivities with 5 mol% catalyst loading (Table 1, entries 2–3). When the organocatalyst 3d was used, the reaction time was 28 h, moderate improvement in enantioselectivity but the lower yield were obtained (Table 1, entry 4 vs. 2–3). The organocatalysts 3c and 3d gives low yield due to absence of hydrogen on the nitrogen atom of amine of the thiourea moiety. When catalyst 3a was used in the reaction, performance of the reaction was better and significant improvement in yield as well as enantioselectivities (Table 1, entry 1 vs. 2–4).
When organocatalyst 3a loading was increased to 8 mol%, it gives moderate yield and enantioselectivity (Table 1, entry 5).Increasing the catalytic loading to 10 mol%, the yield and enantioselectivity of product 6a was improved (Table 1, entry 6). So catalyst 3a (10 mol%) was found to be best in catalytic performance, further we screened different solvents for the domino oxa-Michael-aldol reaction.
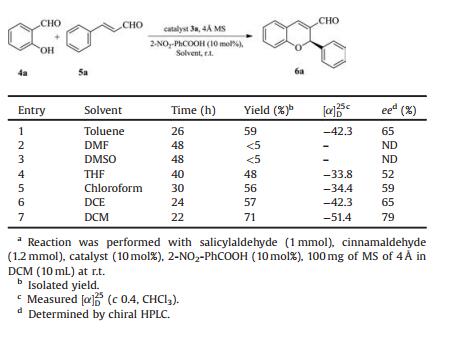
Toluene was reported to provide moderate yield in similar reactions [9], we also observed similar result in our experiment (Table 2, entry 1). This was because the low solubility of catalyst 3a in toluene. When used highly polar solvents such as DMF and DMSO, reaction completed in 48 h giving very poor yields (Table 2, entries 2 and 3), which was in agreement with the literatures [10a, b]. Oxygen containing less polar solvent such as THF gives low yield and poor enantioselectivity (Table 2, entry 4). Less polar halohydrocarbon solvents, such as dichloroethane (DCE) and chloroform, gave moderate yields and enantioselectivities (Table 2, entries 5 and 6). When DCM was used as the solvent, reaction completed in 22 h and gave excellent yield and enantioselectivity. Therefore DCM was chosen as the best solvent (Table 2, entry 7).
|
|
Table 2 Screening of the solvent in domino oxa-Michael-aldol reactiona. |
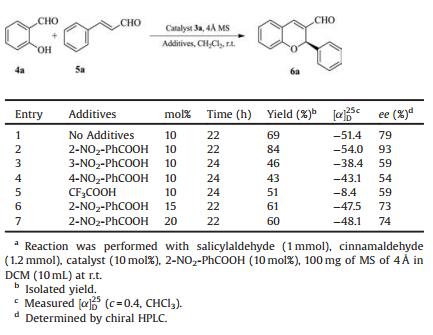
Nevertheless, we investigated the influence of various additives on the organocatalytic oxa-Michael-aldol reaction, and the results were summarized in Table 3. Initially in the absence of any additive we obtained moderate results (Table 3, entry 1). When 2- nitrobenzoic acid (10 mol%) was used, the enantioselectivity of the reaction in DCM was increased from 79% to 93% ee (Table 3, entry 2). A series of nitro substituted benzoic acids were used, such as 3-nitrobenzoic acid and 4-nitrobenzoic acid, we obtained low yields and enantioselectivity (Table 3, entries 3 and 4). When, trifluoroacetic acid (10 mol%) was used, there was no improvement in yield or enantioselectivity for the reaction (Table 3, entry 5). We increased the concentration of 2-nitrobenzoic acid, but the yield and enantioselectivity were decreased (Table 3, entry 2 vs. entries 6 and 7). Eventually, 10 mol% 2-nitrobenzoic acid was identified as the optimal additive.
|
|
Table 3 Effect of the additives on domino oxa-Michael-aldol reactiona. |
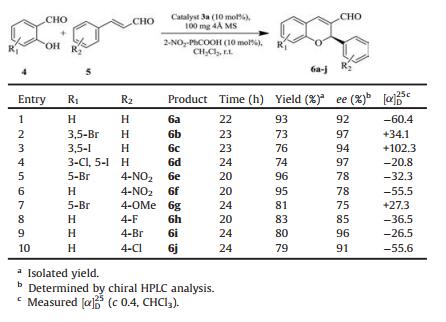
Having established 3a as a suitable organocatalyst, DCM as the solvent and 2-nitrobenzoic acid as an effective additive, then we studied the substrate scope for the enantioselective domino oxaMichael-aldol reaction. The catalytic system worked well for a variety of substituted salicylaldehyde derivatives, although the yields and enantioselectivities varied with the electronic and steric properties of the substrates. The α, β-unsaturated aromatic aldehydes bearing electron-withdrawing groups, such as a nitro group and fluorine were good Michael acceptors, generally affording the desired products 6a–j.
Compound 6a: Isolated yield 93%, [α]D25 -60.1 (c 0.4, CHCl3), 1H NMR (300 MHz, DMSO-d6): δ 5.50 (s, 1H), 6.99-7.05 (d, 2H, J = 7.9, 1.2 Hz), 7.16-7.24 (m, 1H), 7.40-7.45 (t, 2H, J = 7.2, 1.3 Hz), 7.56 (s, 1H), 7.68-7.72 (d, 1H, J = 7.2, 1.3 Hz), 8.07-8.17 (m, 2H), 8.30-8.32 (d, 1H, J = 7.5, 1.1 Hz), 9.46 (s, 1H). 13C NMR (75 MHz, DMSO-d6): δ 74.41, 113.45, 114.34, 118.16, 126.34, 127.89, 129.89, 131.09, 136.49, 142.19, 143.56, 154.43, 160.03, 190.01. HPLC (Daicel Chiralpak AD, hexane/ i-PrOH = 96:4, 0.5 mL/min, 254 nm): tR = 25.36 min (major), 30.45 min (minor).
The enantioselectivity was also affected by the electronic properties of the substituent's of salicylaldehydes as 3, 5-dibromo and 3, 5-diiodo salicylaldehydes showed higher ee than the unsubstituted salicylicaldehyde (Table 4, entries 2–4 and 8–10), while 4-methoxy cinnamaldehyde gives moderate yields and relative lower ee value (Table 4, entry 7).
|
|
Table 4 Asymmetric domino oxa-Michael-aldol reactions of various salicylaldehydes and substituted cinnamaldehydes. |
{kind=link}
In summary, A group of novel proline based organocatalysts derived from Boc-L-proline, 3a-d, have been obtained via a simple synthesis. The catalytic performance of the resultant synthetic products for the domino oxa-Michael-aldol reactions between salicylaldehyde and cinnamaldehyde has been evaluated. It has been found that, out of all four organocatalysts, 3a was found to be efficient for the oxa-Michael-aldol reactions under investigation. It has excellent catalytic activity affording excellent enantioselectivity and moderate to high yields even at low dosages of 10 mol% along with the 2-NO2-PhCOOH (10 mol%) as additives and 100 mg of MS 4 Å.
AcknowledgmentsWe acknowledge, Dr. Smt. S. S. Kadam, Principal and Prof. W. N. Jadhav, Head Department of Chemistry, Dnyanopasak College, Parbhani for providing necessary facilities and University Grant Commission, Delhi for the award of the Junior Research Fellowship.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2017.09.058.
| [1] |
D. Enders, C. Grondal, M.R.M. Hüttl, Angew. Chem. Int. Ed. 46 (2007) 1570-1581. DOI:10.1002/(ISSN)1521-3773 |
| [2] |
E.A. Couladourous, A.T. Strongilos, Angew. Chem. 114 (2002) 3829-3832. DOI:10.1002/1521-3757(20021004)114:19<3829::AID-ANGE3829>3.0.CO;2-1 |
| [3] |
K.C. Nicolaou, J.A. Pfefferkorn, A.J. Roecker, et al., J. Am. Chem. Soc. 122 (2000) 9939-9953. DOI:10.1021/ja002033k |
| [4] |
W.S. Bowers, T. Ohta, J.S. Cleere, P.A. Marsella, Science 193 (1976) 542-547. DOI:10.1126/science.986685 |
| [5] |
(a) P. T. Kaye, M. A. Musa, X. W. Nocada, R. S. Robinson, Org. Biomol. Chem. 1(2003) 1133-1138; (b) P. T. Kaye, M. A. Musa, Synthesis 18(2002) 2701-2706; (c) B. Lesch, S. Brase, Angew. Chem. 116(2004) 118-120; (d) B. Lesch, S. Bruse, Angew. Chem. Int. Ed. 43(2004) 115-118; (e) B. Lesch, J. TorUng, S. Vanderheiden, S. Bruse, Adv. Synth. Catal. 347(2005) 555-562; (f) G. L. Zhao, Y. L. Shi, M. Shi, Org. Lett. 7(2005) 4527-4530. |
| [6] |
(a) H. Pellissier, Tetrahedron 62(2006) 1619-1655; (b) H. Pellissier, Tetrahedron 62(2006) 2143-2173. |
| [7] |
(a) Y. Yamamoto, N. Momiyama, H. Yamamoto, J. Am. Chem. Soc. 126(2004) 5962-5963; (b) M. Marigo, T. Schulte, J. Franzen, K. A. Jorgensen, J. Am. Chem. Soc. 127(2005) 15710-15711; (c) J. W. Yang, M. T. H. Fonseca, B. List, J. Am. Chem. Soc. 127(2005) 15036-15037; (d) Y. Huang, A. M. Walji, C. H. Larsen, J. Am. Chem. Soc. 127(2005) 15051-15053. |
| [8] |
(a) P. B. Thorat, S. V. Goswami, B. C. Khade, S. R. Bhusare, Tetrahedron: Asymmetry 23(2012) 1320-1325; (b) P. B. Thorat, S. V. Goswami, B. C. Khade, S. R. Bhusare, Tetrahedron: Asymmetry 53(2012) 6083-6086; (c) P. B. Thorat, S. V. Goswami, R. L. Magar, B. R. Patil, S. R. Bhusare, Eur. J. Org. Chem. 24(2013) 5509-5516. |
| [9] |
H. Sundén, I. Ibrahem, G.L. Zhao, L. Eriksson, A. Córdova, Chem. Eur J. 13 (2007) 574-581. DOI:10.1002/(ISSN)1521-3765 |
| [10] |
(a) H. Huang, E. N. Jacobsen, J. Am. Chem. Soc. 128(2006) 7170-7171; (b) J. Li, S. Luo, J. Cheng, J. Org. Chem. 74(2009) 1747-1750. |