 2018, Vol. 29
2018, Vol. 29 



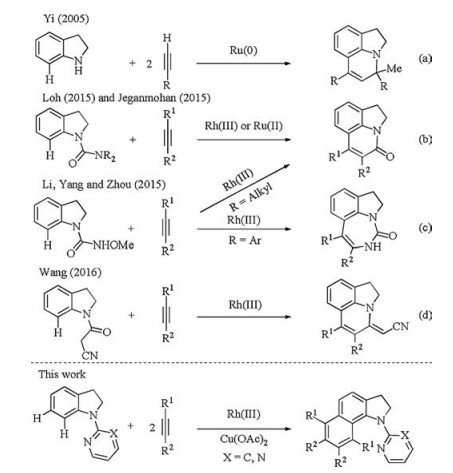
The indole and indoline scaffolds are ubiquitous structural motifs widely found in natural alkaloids, drugs and organic materials [1]. In particular, many pharmaceutical agents include C7-substituted indole and indoline frameworks [2]. Consequently, the development of efficient strategies for regioselective synthesis of indole and indoline derivatives continues to be of great appeal in organic synthesis [3]. Over the past decades, transition-metalcatalyzed direct C—H bond functionalizations have emerged as one of the most efficient synthetic methods to construct diverse organic molecules[4]. In sharp contrast to the vast majority of examples of C—H functionalization of indoles at the C2 and C3 positions[5], similar approaches to access C7-functionalized indoles have very limited reports [6]. However, this type of C—H bond functionalization of indoles can be indirectly achieved by the chelation-assisted selective C7—H bond activation of the indolines, which can be readily converted into the indole moiety by using a suitable oxidizing agent. By employing this strategy, various functionalizations such as arylation [7], alkylation [8], alkenylation [9], alkynylation [10], allylation [11], carbonylation [12], cyanation [13], amination [14] and chalcogenation [15] have been described with the use of various transition-metal catalysts. Equal importantly, the concise synthesis of 1, 7-fused indolines through transition-metal-catalyzed intermolecular direct homologations of indolines with alkynes has been achieved by several groups. As early as in 2005, Yi reported the synthesis of tricyclic quinolones through a cationic ruthenium-hydride complex-catalyzed C7—H bond activation/hydroamination reaction of indolines with alkynes (Scheme 1a) [16]. In 2015, Loh and Jeganmohan independently disclosed the pyrroloquinolinone synthesis by Rh(Ⅲ)- or Ru(Ⅱ)-catalyzed homologation of N-carbamoyl indolines with alkynes through direct C7—H functionalization/C—N cleavage (Scheme 1b) [17, 18]. Li, Yang and Zhou reported a Rh(Ⅲ)-catalyzed redox-neutral C7-selective C—H functionalization of N-methoxycarbamoyl indolines with alkynes, which directly accesses to the seven- as well as sixmembered 1, 7-fused indolines (Scheme 1c) [19]. Recently, Wang and coworkers developed a Rh(Ⅲ)-catalyzed carbocyclization reaction of 3-(indolin-1-yl)-3-oxopropanenitriles with alkynes to form 1, 7-fused indolines (Scheme 1d) [20].

|
Download:
|
| Scheme 1. Rh(Ⅲ)- and Ru(Ⅱ)-catalyzed homologation of indolines with alkynes. | |
The stronger electron-donating ability of the indolinyl nitrogen is likely responsible for the observed C7-reactivity on indoline (electron density C5, C7 > C4, C6). To override this intrinsic siteselectivity, Yu and coworkers have disclosed so far the first and only example of the direct C6—H activation of indolines using a more electron-withdrawing U-shaped nitrile template attached to the indoline via a sulfonamide linkage [21]. However, to the best of our knowledge, there is no report on the catalytic direct both C6- and C7—H bonds functionalization of indolines [22]. Inspired by the pioneering studies and our interest in the transition-metalcatalyzed C—H bond functionalization[23], we herein disclose a facile Rh(Ⅲ)-catalyzed C6- and C7—H indoline homologation with alkynes, in which the N-pyrimidyl group plays a crucial role for this successful transformation (Scheme 1). Moreover, the products obtained could be readily oxidized to the versatile benzo[g]indoles, which are difficult to directly synthesize from indoles due to the inherent C2- and C3-selectivity of indoles in the presence of metal catalysis.
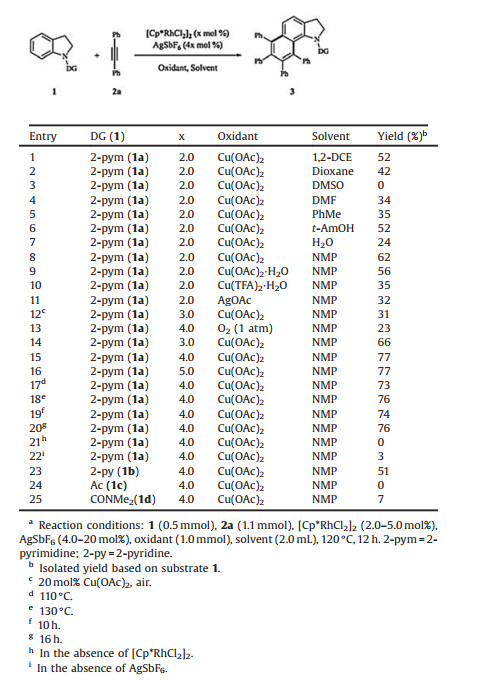
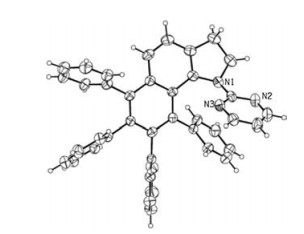
At the outset of our studies, the homologation of N-pyrimidylindoline (1a) with diphenylacetylene (2a) was selected as a model reaction in the presence of [Cp*RhCl2]2 to examine the impact of various parameters as shown in Table 1. The polyarylated benzo[g]indoline 3aa was initially obtained in 52% yield in 1, 2-dichloroethane at 120 ℃ using Cu(OAc)2 and AgSbF6 as the oxidant and additive, respectively (entry 1). The structure of 3aa was confirmed by X-ray single crystal diffraction (Fig. 1). The supplementary crystallographic data of 3aa (CCDC 1817089) can be obtained free of charge from The Cambridge Crystallographic Data Centre. Solvents were evaluated (entries 1–8), and the most efficient catalysis was accomplished in N-methyl pyrrolidone (NMP) (entry 8). Then, oxidants including Cu(OAc)2, Cu(OAc)2·H2O, Cu(TFA)2·H2O, AgOAc as well as O2 were screened (entries 8–13). 2.0 Equivalents of Cu(OAc)2 were suitable for this transformation (62% yield, entry 8). Increasing the loadings of [Cp*RhCl2]2 from 2.0 mol% to 4.0 mol% afforded the desired product 3aa in 77% yield (entries 14–16). Further optimization by decreasing reaction temperature from 120 ℃ to 110 ℃ or increasing to 130 ℃ reduced the yield to some extent (entry 15 vs. entries 17 and 18), and the yield was not evidently improved by prolonging the reaction time (entry 19 vs. entry 20). Moreover, control experiments indicated that no desired product was detected in the absence of [Cp*RhCl2]2 or AgSbF6 (entries 21–22). Then, several popular transition metal catalysts, including Pd(OAc)2, [RuCl2(p-Cymene)]2, [Cp*IrCl2]2 and [Cp*CoI2(CO)] were explored, however, failed to promote this reaction. We examined the coordination efficiency of the N-functionalities to the metal center under the optimal reaction conditions (entry 15). Changing the pyrimidine to pyridine delivered the benzo[g]indoline 3ba in a lower yield (entry 23 vs. entry 15). Whereas carbonyl groups such as -Ac (1c) and -CONMe2 (1d) have been recognized as weekly coordinating directing groups, had a detrimental effect for this current transformation (entries 24–25).
|
|
Table 1 Optimization of various reaction parameters.a |
{kind=link}

|
Download:
|
| Fig. 1. X-ray molecular structure of 3aa. | |
{kind=link}
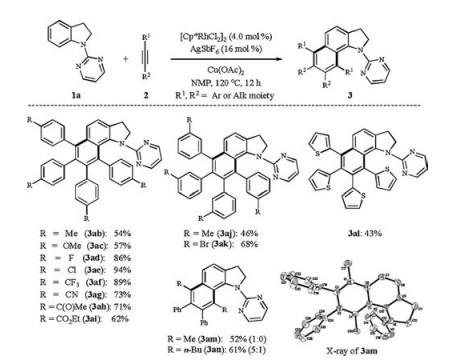
With the optimized reaction condition in hand, various substituted N-pyrimidyl indolines 1 were then subjected to the homologation reaction with diphenylacetylene (2a) (Scheme 2). In general, the methyl and other functional groups (-F, -Cl, -Br, and -CF3) on the aryl moiety were well tolerated, thereby delivering the highly functionalized benzo[g]indolines in moderate to good yields. Concretely, moderate-to-high-yielding transformation of C4-substituted indolines 1e-j was achieved. Among them, indoline 1g bearing electron-withdrawing group (-CF3) reacted efficiently with diphenylacetylene (2a), affording the corresponding annulated product 3ga in 86% yield. The presence of electron-donating groups such as -Me and -OBn on substrates (1e and 1f) caused slightly decreased yields. Indolines 1h-j bearing halo groups were also compatible with this catalytic system, thus offering the opportunity for further transformations in the functionalized products. C5-fluoro or C5-chloro indolines (1k and 1l) underwent the oxidative coupling in a similar manner, affording the corresponding target homologation products 3ka and 3la. Interestingly, the reaction of C5-methyl/C5-methoxyl indolines 1 m/1n with 2a only afforded the hydroarylation product 3 ma/3na probably due to the steric effect from the C5-substituent and rhodium species in the transmetalation step. N-Pyrimidyl indolines 1o-q bearing C2- and/or C3-methyl group(s) proceeded smoothly as well, delivering the corresponding products 3oa-3qa in good yields.

|
Download:
|
| Scheme 2. Oxidative homologation of indolines 1 with diphenylacetylene (2a). | |
{kind=link}
Next, we investigated the scope and limitation of the present dual C—H bond activation of N-pyrimidyl indoline (1a) with various alkynes 2 (Scheme 3). Fluoro (2d), chloro (2e), bromo (2k), trifluoromethyl (2f), cyano (2g) and carbonyl (2h and 2i) groups were well tolerated. Precisely, 1, 2-diarylalkynes (2d-i) with electron-withdrawing substituents worked well, providing the desired products in good to excellent yields, whereas electrondonating groups, such as methyl- (2b) and methoxy- (2c), afforded moderate yields. Besides, substrates 2j and 2k bearing -Me and -Br groups respectively on meta-position of the benzene rings proceeded smoothly to produce the corresponding annulated products 3aj and 3ak in 46% and 68% yields, respectively. However, ortho-methyl or methoxyl group in 1, 2-diarylalkynes had a detrimental effect in the current transformation probably due to the steric hindrance. 1, 2-Di(2-thiophenyl)ethyne (2l) was succesfully subjected to the catalytic system, although only a moderate yield was obtained. To our dissapointment, HPLC analysis indicates only poor regioselectivities for the coupling reaction of indoline 1a with electronically differentiated diaryl-substituted alkynes, such as 1-fluoro-4-(p-tolylethynyl)benzene (for details see the Supporting information). In contrast, unsymmetrical 1, 2-alkylaryl alkynes 2m-n could proceed smoothly with high regioselectivity under the rhodium catalytic system, clearly favoring an orientation of the aromatic substituents at the 2, 3-position over the 2, 4-position. The structure and regiochemistry of compound 3am was confirmed by X-ray single crystal diffraction. The supplementary crystallographic data CCDC 1, 817, 090 can be obtained free of charge from the Cambridge Crystallographic Data Centre. When aliphatic alkyne 5-decyne as well as termical alkyne ethynylbenzene were tested under the standard conditions, only a trace amount of products was observed.

|
Download:
|
| Scheme 3. Oxidative homologation of indoline 1a with substituted alkynes 2. | |
{kind=link}
Given the high catalytic efficacy of the Rh(Ⅲ)-catalyzeddualC—H activation reaction, we became interested in probing its mode of action. Intermolecular competition experiments between indolines and alkynes rendered electrophilic-type C—H bond activation in the homologation reaction. The oxidative homologation of N-pyrimidyl indoline (1a) with diphenylacetylene (2a) in the presence of [D]1- AcOH under the standard reaction conditions was conducted. Significant deuterium was observed from the recycled substrate [D]n-1a(30% D), being indicative of a reversible C—H bond metalation step (for details see the Supporting information).
Finally, we investigated the possibility to the further synthetic utility of the benzo[g]indoline products (Scheme 4) [15b]. Delightedly, treatment of 3aa with DDQ generated N-pyrimidyl benzo[g]indole 4aa in 76% yield, which further furnished NH-free benzo[g]indole 5aa in moderate yield under basic hydrolysis conditions.

|
Download:
|
| Scheme 4. Transformation of benzo[g]indoline 3aa. | |
{kind=link}
In conclusion, we have developed a rhodium-catalyzed oxidative homologation of N-pyrimidyl indolines with alkynes through dual C—H bond activation. The balance between the directing ability and the electrophilicity of the pyrimidyl group enables the homologation to proceed successfully. The homologation reaction was compatible with various functional indolines and symmetrical as well as unsymmetrical alkyl-aryl alkynes with high regioselectivity. Moreover, the benzo[g]indoline products obtained could be readily oxidized to the versatile benzo[g]indoles, which are difficult to directly synthesize from indoles.
AcknowledgmentsThis research was supported by the National Natural Science Foundation of China (Nos. 21602064 and 21572072), the Natural Science Foundation of Fujian Province (No. 2015J01056), and Huaqiao University (No. ZQN-PY317).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.05.007.
| [1] |
(a) W. Zi, Z. Zuo, D. Ma, Acc. Chem. Res. 48(2015) 702-711; (b) W. Xu, D. J. Gavia, Y. Tang, Nat. Prod. Rep. 31(2014) 1474-1487. |
| [2] |
J.P. Burke, Z. Bian, S. Shaw, et al., J. Med. Chem. 58 (2015) 3794-3805. DOI:10.1021/jm501984f |
| [3] |
(a) S. Cacchi, G. Fabrizi, Chem. Rev. 111(2011) PR215-PR283; (b) G. Bartoli, G. Bencivenni, R. Dalpozzo, Chem. Soc. Rev. 39(2010) 4449-4465. |
| [4] |
(a) Y. Hu, B. Zhou, C. Wang, Acc. Chem. Res. 51(2018) 816-827; (b) Special issue on CH activation, Chem. Rev. 117(2017) 8481-9520; (c) J. K. Kim, K. Shin, S. Chang, Top. Organomet. Chem. 55(2016) 29-51; (d) W. Liu, L. Ackermann, ACS Catal. 6(2016) 3743-3752; (e) L. Yang, H. Huang, Chem. Rev. 115(2015) 3468-3517; (f) Z. Wang, P. Xie, Y. Xia, Chin. Chem. Lett. 29(2018) 47-53; (g) R. Y. Zhu, Z. Q. Li, H. S. Park, C. H. Senanayake, J. Q. Yu, J. Am. Chem. Soc. 140(2018) 3564-3568; (h) N. Sauermann, R. Mei, L. Ackermann, Angew. Chem. Int. Ed. 57(2018) 5090-5094; (i) Z. J. Cai, C. X. Liu, Q. Gu, S. L. You, Angew. Chem. Int. Ed. 57(2018) 1296-1299; (j) B. H. Zhang, J. J. Kong, Y. Huang, et al., Chin. Chem. Lett. 28(2017) 1751-1754. |
| [5] |
(a) A. H. Sandtorv, Adv. Synth. Catal. 357(2015) 2403-2435; (b) E. M. Beck, M. J. Gaunt, Top. Curr. Chem. 292(2010) 85-121. |
| [6] |
J.A. Leitch, Y. Bhonoah, C.G. Frost, ACS Catal. 7 (2017) 5618-5627. DOI:10.1021/acscatal.7b01785 |
| [7] |
(a) Z. Shi, B. Li, X. Wan, et al., Angew. Chem. Int. Ed. 46(2007) 5554-5558; (b) T. Nishikata, A. R. Abela, S. Huang, B. H. Lipshutz, J. Am. Chem. Soc. 132(2010) 4978-4979. |
| [8] |
(a) C. Premi, A. Dixit, N. Jain, Org. Lett. 17(2015) 2598-2601; (b) H. Jo, J. Park, M. Choi, et al., Adv. Synth. Catal. 358(2016) 2714-2720. |
| [9] |
(a) B. Urones, R. G. Arrayás, J. C. Carretero, Org. Lett. 15(2013) 1120-1123; (b) X. F. Yang, X. H. Hu, C. Feng, T. P. Loh, Chem. Commun. 51(2015) 2532-2535. |
| [10] |
(a) N. Jin, C. Pan, H. Zhang, et al., Adv. Synth. Catal. 357(2015) 1149-1153; (b) Y. Wu, Y. Yang, B. Zhou, Y. Li, J. Org. Chem. 80(2015) 1946-1951. |
| [11] |
M. Choi, J. Park, S. Sharma, et al., J. Org. Chem. 81 (2016) 4771-4778. DOI:10.1021/acs.joc.6b00721 |
| [12] |
(a) P. L. Wang, Y. Li, L. Ma, et al., Adv. Synth. Catal. 358(2016) 1048-1053; (b) T. Jeong, S. H. Lee, N. K. Mishra, et al., Adv. Synth. Catal. 359(2017) 2329-2336. |
| [13] |
N.K. Mishra, T. Jeong, S. Sharma, et al., Adv. Synth. Catal. 357 (2015) 1293-1298. DOI:10.1002/adsc.201401152 |
| [14] |
(a) C. Pan, A. Abdukader, J. Han, Y. Cheng, C. Zhu, Chem. Eur. J. 20(2014) 3606-3609; (b) K. Shin, S. Chang, J. Org. Chem. 79(2014) 12197-12204. |
| [15] |
(a) W. Xie, B. Li, B. Wang, J. Org. Chem. 81(2016) 396-403; (b) P. Gandeepan, J. Koeller, L. Ackermann, ACS Catal. 7(2017) 1030-1034. |
| [16] |
C.S. Yi, S.Y. Yun, I.A. Guzei, J. Am. Chem. Soc. 127 (2005) 5782-5783. DOI:10.1021/ja042318n |
| [17] |
X.F. Yang, X.H. Hu, T.P. Loh, Org. Lett. 17 (2015) 1481-1484. DOI:10.1021/acs.orglett.5b00355 |
| [18] |
R. Manoharan, M. Jeganmohan, Org. Biomol. Chem. 13 (2015) 9276-9284. DOI:10.1039/C5OB01146A |
| [19] |
X. Wang, H. Tang, H. Feng, et al., J. Org. Chem. 80 (2015) 6238-6249. DOI:10.1021/acs.joc.5b00684 |
| [20] |
T. Zhou, Y. Wang, B. Li, B. Wang, Org. Lett. 18 (2016) 5066-5069. DOI:10.1021/acs.orglett.6b02521 |
| [21] |
G. Yang, P. Lindovska, D. Zhu, et al., J. Am. Chem. Soc. 136 (2014) 10807-10813. DOI:10.1021/ja505737x |
| [22] |
(a) T. Satoh, M. Miura, Chem. Eur. J. 16(2010) 11212-11222; (b) J. Zheng, S. L. You, Chem. Commun. 50(2014) 8204-8207; (c) B. Liu, F. Hu, B. F. Shi, Adv. Synth. Catal. 356(2014) 2688-2696; (d) X. Tan, B. Liu, X. Li, et al., J. Am. Chem. Soc. 134(2012) 16163-16166; (e) Z. Shi, C. Tang, N. Jiao, Adv. Synth. Catal. 354(2012) 2695-2700; (f) G. Song, X. Gong, X. Li, J. Org. Chem. 76(2011) 7583-7589; (g) N. Umeda, H. Tsurugi, T. Satoh, M. Miura, Angew. Chem. Int. Ed. 47(2008) 4019-4022; (h) Y. Wang, H. Zhou, K. Xu, M. H. Shen, H. D. Xu, Chin. Chem. Lett. 28(2017) 92-96. |
| [23] |
(a) L. Wang, Y. Yu, M. Yang, et al., Adv. Synth. Catal. 359(2017) 3818-3825; (b) J. Wu, X. Cui, X. Mi, Y. Li, Y. Wu, Chem. Commun. 46(2010) 6771-6773. |