 2018, Vol. 29
2018, Vol. 29 



b Advanced Materials and Catalysis Group, ZJU-NHU United R&D Center, Department of Chemistry, Zhejiang University, Hangzhou 310028, China
Cyclohexanol is an important intermediate for the synthesis of Nylon-6 and as well as the component of KA oil using in petroleum industrial chemistry [1, 2]. In addition, the catalytic transformation of phenols to stable hydrocarbons is of great significance in the upgrading of bio-oil [3]. At present, several catalytic systems have been successfully applied into the transformation, such as the selective oxidation of cyclohexane [4, 5], direct hydration of cyclohexene [6] and the hydrogenation of phenol [7, 8]. For selective oxidation of cyclohexane and direct hydration of cyclohexene, more or less drawbacks existed, such as complicated steps, explosion risk, low conversion and selectivity [9]. Among these processes, the hydrogenation of phenol was considered to be the most efficient and promising strategy considering the high atom utilization and the green process. What is more, phenol is readily available from naturally lignins, which is one of the most abundant available feedstocks [10]. The wide sources of raw materials guarantee the reaction take place smoothly and provide the possibility for industrial applications.
Up to now, a series of studies have been reported for the hydrogenation of phenols. Particularly, heterogeneous catalysts have received an astonishing amount of attention due to their easy recovery and good reusability. The active metals play an important role in the selective hydrogenation reactions. Specifically, the adsorption and dissociation of hydrogen and the adsorption behavior of substrate on active metals are directly related to the catalytic activity and selectivity [11]. The present catalysts used in the catalytic hydrogenation of phenols are mainly precious metals, especially Pd [12-22], Pt [23, 24], Rh [25, 26] and Ru [27-29]. Although the precious metals catalysts showed good catalytic activity and selectivity to cyclohexanone or cyclohexanol, the risk in supply and volatile price of noble metals inhibited to accomplish industrializaiton. To circumvent the problems, development of catalysts based on earth-abundant metals, such as Fe, Co, and Ni, was regarded as an impressive strategy considering their distinct electronic structures [30] and low cost. Very recently, important progress made in the fabrication of earth-abundant metal catalysts using in chemoselective hydrogenation reactions [31-36], oxidation reactions [37-39] and electrocatalytic reactions [40-42]. However, the hydrogenation of phenol using non-noble metal catalysts is rare reported [7, 43] and should be emphasized. It is worth mentioning that base metals are easy to be oxidized and cause irreversible deactivation of the catalysts under liquid-phase conditions. Therefore, it is necessary to develop a convenient strategy for the synthesis of stable, high activity and selectivity non-noble metal catalysts.
Porous carbon materials are fascinating materials that have attracted widespread attention due to the high surface area, rich pore sturcture, high stability and so on. They provide access to a wide range of applications, especially act as good supports for heterogeneous catalysts [44, 45]. The introduction of porous carbon can stable the nanoparticles (NPs) and enhance the interaction between the supports and NPs, thus benefiting the catalytic process. Herein, we effectively combine porous carbon and cobalt oxides by direct pyrolysis of D-glucosamine hydrochloride (GAH), melamine and CoCl2·6H2O. The obtained hybrids (CoOx@CN) show high activity and selectivity in the hydrogenation of phenol to cyclohexanol. Almost full conversion (98%) of phenol with excellent selectivity (> 99%) toward cyclohexanol were obtained under optimized reaction conditions. Detailed analysis of the relationship between catalytic performance and catalyst component indicates that the synergistic effect occurs during the raction. In particular, the original Co3O4 in CoOx@CN is benificial for the adsorption and activation of phenol and the in situ gernerated Co is responsible for hydrogen adsorption and dissociation, which greatly improve the catalytic activity. The successful integration of porous carbon and earth-abundant metals in the new hybrid paves the way for design of more intriguing materials in heterogeneous catalysis.
The hybrids (CoOx@CN) were prepared in one step through pyrolysis of GAH, melamine and CoCl2·6H2O at 800 ℃. For comparasion, CoCl2/AC and CoCl2/commercial-CNTs were synthesized under the identical conditions, and the resultants were named CoOx@AC and CoOx@CNT, respectively. To access the microstructures of CoOx@CN, a series of characterizations, such as transmission electron microscopy (TEM), N2 adsorption/ desorption isotherms, X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) were conducted. As shown in Fig. S1a (Supporting information), the N2 adsorption/desorption isotherm resembled type Ⅳ with a hysteresis loop, indicating the existence of mesopores. The calculated specific surface area (SSA) was 344 m2/g and the mean pore size was 10 nm. Such large SSA and wide pore diameter were beneficial to expose more active sites and mass transfer. In the XRD pattern of CoOx@CN (Fig. S1b in Supporting information), the diffraction peak at 26° was ascribed to the (002) reflection of the graphitic-type lattice. The diffraction peaks at 19°, 31.2°, 36.8°, 44.8°, 55.7°, 59.4°, 65.2° could be assigned to the (111), (220), (311), (400), (422), (511), (440) planes of Co3O4 nanocrystal (PDF 42-1467). Besides, a weak reflection at 61.5° was the characteristic (220) plane of CoO. Therefore, Co3O4 is the main phase in CoOx@CN. TEM images further revealed the microstructure of CoOx@CN. In Fig. S1c in Supporting information, the carbon support consisted of multi-walled carbon nanotubes and graphite layers. The CoOx NPs dispersed uniformly on the carbon support with a mean size of 13.8 nm by counting > 300 metal NPs. The ICPAES test demonstrated that the content of cobalt was 27 wt%.
To evaluate the catalytic performance of CoOx@CN, we select the hydrogenation of the benchmark substrate phenol as a model reaction. The reaction conditions were optimized through variation of the solvent, temperature, and hydrogen pressure. First, we assessed the effects of solvent on the reaction. Among the tested solvents listed in Table S1 (Supporting information), water gave the best result, furnishing a satisfying yield (98%) toward cyclohexanol, which was economically viable because raw bio-oil contains 10–13 wt% water. Ethanol and i-propanol also performed moderate catalytic activity and perfect selectivity. In contrast, completely no conversion was obtained in THF, dimethyl sulfoxide and dioxane. It seems that the reaction rate is not correlated with solvent polarity or solubility of H2. It should be noted that CoOx@CN performed much better activity in protic solvent, while CoOx@CN had no activity in nonprotic solvents at all. This phenomenon was consistent with the conclusion reported by Lercher [46]. Protic solvents can provide proton and form hydrogen bond with phenol, which can lower the activation energy of the reaction and promote the hydrogenation process [47, 48].
Additionally, the temperature and hydrogen pressure affected the performance of catalysts enormously. As shown in Table S2 (Supporting information), the catalytic system is sensitive to temperature and hydrogen pressure, especially the conversion of phenol. At 120 ℃ and 3 MPa H2, the conversion of phenol was 43% and the selectivity was perfect. Elevating the temperature to 135 ℃ with constant pressure, the conversion increased to 62%. Pleasingly, furthur raising the reaction temperature to 150 ℃ afforded an excellent conversion, furnishing cyclohexanol in 98% yield. At 150 ℃, decreasing the hydrogen pressure from 3 MPa to 2.5 MPa, the conversion of phenol declined markedly to 35% yet excellent selectivity. Further reducing the hydrogen pressure to 2 MPa led to the worst conversion, obtaining only 17% yield of the target product. For follow-up studies, we conducted the reaction at 150 ℃ and 3 MPa, as these conditions represented the best performance of CoOx@CN.
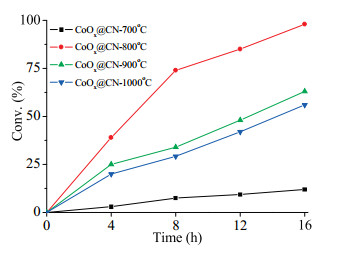
As the cobalt-based hybrids were prepared under high temperature, different pyrolysis temperature can affect the structure and composition of the catalyst, thus influencing its catalytic activity. We then investigated the performance of the catalysts pyrolysis from 700 ℃ to 1000 ℃ under optimized reaction conditions (150 ℃, 3 MPa H2). Evolution of conversion with reaction time by a series of catalysts were conducted and the results were compiled in Fig. 1. We found that the catalysts pyrolyzed at different temperature behaved differently. On increasing the pyrolysis temperature from 700 ℃ to 1000 ℃, the activity of the resulting catalyst increased firstly and then decreased. The catalytic activity of CoOx@CN-700 ℃ was relatively weak, affording only 12% yield of cyclohexanol. While the material pyrolyzed at 800 ℃ exhibited the highest catalytic activity, producing cyclohexanol in 98% yield.

|
Download:
|
| Fig. 1. Evolution of conversion with reaction time by CoOx@CN under different pyrolysis temperature. | |
In order to study the reasons for the difference in catalytic activity, a series of characterizations were performed. As the valence state of active metal is closely related to the catalytic activity, the species of cobalt should be discussed. To this end, we used XPS and XRD to access the cobalt composition in different catalysts (Fig. S2 in Supporting information). The XPS results showed that the valence state of Co in different catalysts was almost the same. The peaks located at 780 eV and 795.6 eV in the spectrum were corresponding to the Co3O4 phase, and the characteristic peak of the metallic Co was not found. Therefore, Co3O4 was the main phase in the tested catalysts, which was also verified by the XRD results (Fig. S2d). The similar cobalt species in the catalysts pyrolyzed at different temperature is not possible to explain the reason of the difference in the activity.
As we know, the SSA and pore structure can affect mass transfer during the reaction. Next, we applied N2 adsorption/desorption to analysis the textural properties of different catalysts. As displayed in Fig. S3 (Supporting information), the catalyst calcinated at 700 ℃ exhibited the relatively small SSA and pore volume, which was attributed to that the template agent (carbon nitride) was not completely decomposed under this temperature. Correspondingly, CoOx@CN-700 ℃ showed the worst activity. High temperature annealling (> 700 ℃) lead to the complete decomposion of template agent, during which the produced small molecule can activate the hybrid and improve the SSA and pore volume. Among the tested catalysts, we found that the material pyrolyzed at 800 ℃ (CoOx@CN) possessed the maximum SSA (344 m2/g) with the corresponding highest conversion. That is to say, the textural structure of CoOx@CN had an important influence on the catalytic activity.
Apart from the textural structure, the dispersion and the mean particle size of metal NPs greatly affected the performance of catalysts. Here, we employed TEM images to obtain the mean particle size and studied the relationship between the catalytic activity and the men particle size. The metal particle size distribution histogram in Fig. S4 (Supporting information) showed that CoOx@CN-800 ℃ and CoOx@CN-900 ℃ had the similar metal distribution and the mean particle size was around 13 nm. However, raising the pyrolysis temperature to 1000 ℃, the agglomeration of NPs occurred and the mean particle size increased to 19.2 nm rapidly (Fig. S4c). It can be concluded that the growth of NPs caused the decresed acticity. By a thorough analysis, we have reason to believe that the high SSA and the homogeneous dispersion of NPs jointly promoted the reaction.
CoOx@CN exhibited the highest catalytic activity in the hydrogenation of phenol. Actually, metallic cobalt (Co) can activate hydrogen and act as the real active sites in the chemoselective hydrogenation of nitroarenes [32] and quinoline compounds [49]. However, Co3O4 was the main metal phase in CoOx@CN, which cannot activate hydrogen. It was necessary to uncover the real active sites in the hydrogenation of phenol. For comparison, the catalytic activities of commercial Co3O4, CoO and Co were examined. Co and CoO catalyst showed similar activity, giving indeed low yield of 16% and 17%, respectively (Table 1, entries 1–2). In contrast, the commercial Co3O4 showed significantly increased activity, acquiring 75% yield of cyclohexanol (Table 1, entry 3). It seems that Co3O4 NPs may be real active sites.
|
|
Table 1 Catalytic hydrogenation of phenol using different Co catalystsa. |

{kind=link}
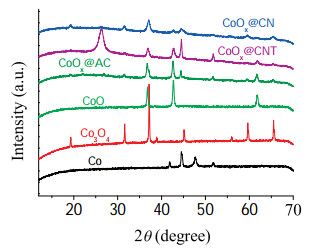
To deeply gain insight into the catalytic active sites, a series of control experiments were conducted. As shown in Table 3, CoOx@CNTand CoOx@AC obtained negligible yield of cyclohexanol. The XRD profiles of different catalysts in Fig. 2 demonstrated that the main metal phases in CoOx@CNT and CoOx@AC were Co and CoO, while the characteristic diffraction of Co3O4 was not found. By comparison with the catalyst components and the activity of CoOx@CN, it again suggested that Co3O4 gave a big boost to the performance of the catalyst.

|
Download:
|
| Fig. 2. XRD profiles of different catalysts. | |
{kind=link}
In order to figure out the role of Co3O4, we monitor the chemical reaction course at 150 ℃ and 3 MPa. Taking the reaction time as the X-axis, the conversion as the Y-axis, we draw the diagram shown in Fig. 3. In the initial stage of the reaction, the conversion of phenol remained at a very low level. After an induction period, the conversion steadily increased as the reaction time prolonged to 24 h. It is worth mentioning that the reaction rate increased first and then decreased. The volcano curve of reaction rate indicated that the Co3O4 phase in CoOx@CN may change during the hydrogenation of phenol. To verify the hypothesis, we used XRD to explore the variation of valence state in CoOx@CN during the reaction.

|
Download:
|
| Fig. 3. The time-activity profile for hydrogenation of phenol under Co3O4 catalyst and its differential function. | |
{kind=link}
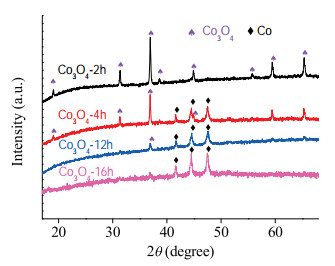
The XRD results in Fig. 4 indicated that at the initial stage of the reaction with low conversion of phenol, the diffraction peaks of Co3O4 were recorded and Co was not formed. Note that Co3O4 was gradually reduced to Co as the reaction progress. Correspondingly, the conversion of phenol increased dramatically. When Co3O4 was completely reduced to Co, the conversion tended to be stable. It was reasonable to believe that Co3O4 in CoOx@CN will also be in situ reduced to cobalt during the hydrogenation process. To illustrate this, hydrogen temperature-programmed reduction (H2-TPR) was employed to analyze the interaction between the catalyst and hydrogen.

|
Download:
|
| Fig. 4. XRD profiles of Co3O4 under different reaction time. | |
{kind=link}
By comparison the H2-TPR spectrum of Co3O4, Co and CoOx@CN (Fig. S5 in Supporting information), we found that two representative reduction peaks of Co3O4 (Co3O4-CoO, CoO-Co) were recorded in CoOx@CN and the starting reduction temperature was 184 ℃. However, the commercial Co3O4 started to be reduced at 301 ℃, which was extraordinarily higher than the Co3O4 in CoOx@CN. It suggested that the Co3O4 in CoOx@CN was readily reduced to Co under the reaction conditions, which can be attributed to the homogeneous dispersion of NPs and the interaction between NPs and porous carbon. The introduction of porous carbon can disperse NPs and make Co3O4 easy to be reduced to Co, thus producing more active sites and accelerating the reaction.
Based on the above discussion, it demonstrated that Co NPs were the active components in CoOx@CN, which dissociated H2 and formed activated-H species. Chen and co-workers recently reported that benzene was adsorbed and activated at Co3O4 sites via the interaction between its π-electrons and the positively charged sites (holes) of Co3O4 [50, 51]. The group of Nelson also disclosed that the adsorption and activation of phenol occurred on oxide support (CeO2) [52]. Therefore, in our catalytic system, the original Co3O4 in CoOx@CN was responsible for the adsorption and activation of phenol and the in situ generated Co provided hydrogen adsorption and dissociation sites, thus improving the catalytic activity. The synergetic effect between Co3O4 and Co was absolutely vital for the high catalytic activity of CoOx@CN. To provide more evidence about the synergetic effect of Co and Co3O4, we conducted the experimrnts at 160 ℃. 80% yiled of cyclohexanol was obtained in 12 h uing Co3O4, higher than the activity at 150 ℃. By a detailed analysis of the metal phase with XRD, we found that most of Co3O4 had been converted to Co0 in 8 h at 160 ℃. Prolonging the reaction time to 12 h, all the Co3O4 had been reduced to Co0 (Fig. S6 in Supporting information). By contrast, the totally transformation from Co3O4 to Co0 needed 16 h as shown in Fig. 4. That is, the higher reaction temperature was benificial to the in situ reduction of Co3O4 to Co0, promoted the formation of Co3O4-Co0 dual active sites, thus improving the hydrogenation of phenol.
In summary, we developed a cheap and convenient strategy for the fabrication of cobalt oxide and porous carbon hybrids (CoOx@CN) starting from biomass, CoCl2·6H2O and melamine. The obtained materials were successfully applied into the chemoselective hydrogenation of phenol into cyclohexanol. The high activity and selectivity of CoOx@CN was attributed to high SSA and the homogeneously dispersed NPs. More importantly, the synergistic effect between Co3O4 and the in situ generated Co during the reaction greatly promoted the reaction. In detail, the Co3O4 in CoOx@CN played an important role for the adsorption and activation of phenol and the in situ generated Co was responsible for hydrogen adsorption and dissociation. These findings open new avenues for the development of low-cost catalysts in industrially relevant processes.
AcknowledgmentsFinancial support from the key program supported by the Natural Science Foundation of Zhejiang Province, China (No. LZ18B060002), the National Natural Science Foundation of China (No. 21622308), the Specialized Research Fund for the Doctoral Program of Higher Education (No. J20130060), the Fundamental Research Funds for the Central Universities, and the Program for Zhejiang Leading Team of S&T Innovation are greatly appreciated.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.01.020.
| [1] |
U. Schuchardt, D. Cardoso, R. Sercheli, et al., Appl. Catal. A-Gen. 211 (2001) 1-17. DOI:10.1016/S0926-860X(01)00472-0 |
| [2] |
J. Long, S. Shu, Q. Wu, et al., Energy Convers. Manage. 105 (2015) 570-577. |
| [3] |
J.O. Metzger, A. Hüttermann, Naturwissenschaften 96 (2009) 279-288. DOI:10.1007/s00114-008-0479-4 |
| [4] |
Y. Liu, H. Tsunoyama, T. Akita, S. Xie, T. Tsukuda, ACS Catal. 1 (2011) 2-6. DOI:10.1021/cs100043j |
| [5] |
X. Fang, Z. Yin, H. Wang, et al., J. Catal. 329 (2015) 187-194. DOI:10.1016/j.jcat.2015.05.004 |
| [6] |
B.C. Chen, B.Y. Yu, Y.L. Lin, H.P. Huang, I.L. Chien, Ind. Eng. Chem. Res. 53 (2014) 7079-7086. DOI:10.1021/ie404226m |
| [7] |
A.Q. Li, K. Shen, J.Y. Chen, Z. Li, Y.W. Li, Chem. Eng. Sci. 166 (2017) 66-76. DOI:10.1016/j.ces.2017.03.027 |
| [8] |
J. Long, S. Shu, Q. Wu, et al., Energy Convers. Manage. 105 (2015) 570-577. DOI:10.1016/j.enconman.2015.08.016 |
| [9] |
L. Zhou, J. Xu, H. Miao, F. Wang, X. Li, Appl. Catal. A-Gen. 292 (2005) 223-228. DOI:10.1016/j.apcata.2005.06.018 |
| [10] |
X.J. Cui, A.E. Surkus, K. Junge, et al., Nat. Commun. 7 (2016) 11326. DOI:10.1038/ncomms11326 |
| [11] |
N. Mahata, V. Vishwanathan, Adsorpt. Sci. Technol. 15 (1997) 165-172. DOI:10.1177/026361749701500303 |
| [12] |
H.Z. Liu, T. Jiang, B.X. Han, S.G. Liang, Y.X. Zhou, Science 326 (2009) 1250-1252. DOI:10.1126/science.1179713 |
| [13] |
P. Claus, H. Berndt, C. Mohr, et al., J. Catal. 192 (2000) 88-97. DOI:10.1006/jcat.2000.2834 |
| [14] |
J.F. Zhu, G.H. Tao, H.Y. Liu, et al., Green Chem. 16 (2014) 2664-2669. DOI:10.1039/C3GC42408A |
| [15] |
E. Diaz, A.F. Mohedano, L. Calvo, et al., Chem. Eng. J. 131 (2007) 65-71. DOI:10.1016/j.cej.2006.12.020 |
| [16] |
Y. Wang, J. Yao, H.R. Li, D.S. Su, M. Antonietti, J. Am. Chem. Soc. 133 (2011) 2362-2365. DOI:10.1021/ja109856y |
| [17] |
Z.L. Li, J.H. Liu, C.G. Xia, F.W. Li, ACS Catal. 3 (2013) 2440-2448. DOI:10.1021/cs400506q |
| [18] |
G.F. Li, J.Y. Han, H. Wang, X.L. Zhu, Q.F. Ge, ACS Catal. 5 (2015) 2009-2016. DOI:10.1021/cs501805y |
| [19] |
N.C. Nelson, J.S. Manzano, A.D. Sadow, S.H. Overbury, I.I. Sowing, ACS Catal. 5 (2015) 2051-2061. DOI:10.1021/cs502000j |
| [20] |
Y.Z. Chen, G.R. Cai, Y.M. Wang, et al., Green Chem. 18 (2016) 1212-1217. DOI:10.1039/C5GC02530C |
| [21] |
H.L. Liu, Y.W. Li, R. Luque, H.F. Jiang, Adv. Synth. Catal. 353 (2011) 3107-3113. DOI:10.1002/adsc.v353.17 |
| [22] |
H.F. Li, Q.S. Zhang, Z.B. Pang, et al., Chin. Chem. Lett. 27 (2016) 1500-1504. DOI:10.1016/j.cclet.2016.03.036 |
| [23] |
X. Yang, X. Yu, L.Z. Long, et al., Chem. Commun. 50 (2014) 2794-2796. DOI:10.1039/c3cc49331h |
| [24] |
B. Sarkar, C. Pendem, L.N.S. Konathala, T. Sasaki, R. Bal, J. Mater. Chem. A 2 (2014) 18398-18404. DOI:10.1039/C4TA04542D |
| [25] |
I.E. Ertas, M. Gulcan, A. Bulut, M. Yurderi, M. Zahmakiran, J. Mol. Catal. A:Chem. 410 (2015) 209-220. DOI:10.1016/j.molcata.2015.09.025 |
| [26] |
S. Kuklin, A. Maximov, A. Zolotukhina, E. Karakhanov, Catal. Commun. 73 (2016) 63-68. DOI:10.1016/j.catcom.2015.10.005 |
| [27] |
F. Lu, J. Liu, J. Xu, Mater. Chem. Phys. 108 (2008) 369-374. DOI:10.1016/j.matchemphys.2007.10.010 |
| [28] |
I.E. Ertas, M. Gulcan, A. Bulut, M. Yurderi, M. Zahmakiran, Microporous Mesoporous Mater. 226 (2016) 94-103. DOI:10.1016/j.micromeso.2015.12.048 |
| [29] |
A. Maximov, A. Zolotukhina, V. Murzin, E. Karakhanov, E. Rosenberg, ChemCatChem 7 (2015) 1197-1210. DOI:10.1002/cctc.201403054 |
| [30] |
M.R. Friedfeld, M. Shevlin, J.M. Hoyt, et al., Science 342 (2013) 1076-1080. DOI:10.1126/science.1243550 |
| [31] |
R.V. Jagadeesh, A. E. Surkus, H. Junge, et al., Science 342 (2013) 1073-1076. DOI:10.1126/science.1242005 |
| [32] |
Z.Z. Wei, J. Wang, S.J. Mao, et al., ACS Catal. 5 (2015) 4783-4789. DOI:10.1021/acscatal.5b00737 |
| [33] |
F.A. Westerhaus, R.V. Jagadeesh, G. Wienhofer, et al., Nat. Chem. 5 (2013) 537-543. DOI:10.1038/nchem.1645 |
| [34] |
T. Schwob, R. Kempe, Angew. Chem. Int. Ed. 55 (2016) 15175-15179. DOI:10.1002/anie.201608321 |
| [35] |
F.W. Zhang, C. Zhao, S. Chen, et al., J. Catal. 348 (2017) 212-222. DOI:10.1016/j.jcat.2017.02.028 |
| [36] |
P.F. Ji, K. Manna, Z. Lin, et al., J. Am. Chem. Soc. 139 (2017) 7004-7011. DOI:10.1021/jacs.7b02394 |
| [37] |
D. Banerjee, R.V. Jagadeesh, K. Junge, et al., Angew. Chem. Int. Ed. 53 (2014) 4359-4363. DOI:10.1002/anie.201310420 |
| [38] |
K. Shen, X. Chen, J. Chen, Y. Li, ACS Catal. 6 (2016) 5887-5903. DOI:10.1021/acscatal.6b01222 |
| [39] |
W. Zhong, H. Liu, C. Bai, S. Liao, Y. Li, ACS Catal. 5 (2015) 1850-1856. DOI:10.1021/cs502101c |
| [40] |
H.Y. Jin, J. Wang, D.F. Su, et al., J. Am. Chem. Soc. 137 (2015) 2688-2694. DOI:10.1021/ja5127165 |
| [41] |
J. Wang, S. Mao, Z. Liu, et al., ACS Appl. Mater. Interfaces 9 (2017) 7139-7147. DOI:10.1021/acsami.6b15377 |
| [42] |
J. Wang, Z. Wei, H. Wang, Y. Chen, Y. Wang, J. Mater. Chem. A 5 (2017) 10510-10516. DOI:10.1039/C7TA02115A |
| [43] |
Y. Xiang, X. Li, C. Lu, et al., Ind. Eng. Chem. Res. 50 (2011) 3139-3144. DOI:10.1021/ie101411h |
| [44] |
X. Xu, Y. Li, Y. Gong, et al., J. Am. Chem. Soc. 134 (2012) 16987-16990. DOI:10.1021/ja308139s |
| [45] |
P.F. Zhang, Y.T. Gong, H.R. Li, Z.R. Chen, Y. Wang, Nat. Commun. 4 (2013) 1593. DOI:10.1038/ncomms2586 |
| [46] |
J. He, C. Zhao, J.A. Lercher, J. Catal. 309 (2014) 362-375. DOI:10.1016/j.jcat.2013.09.009 |
| [47] |
Y. Xiang, L. Kong, P. Xie, et al., Ind. Eng. Chem. Res. 53 (2014) 2197-2203. DOI:10.1021/ie4035253 |
| [48] |
Y. Yoon, R. Rousseau, R.S. Weber, D. Mei, J.A. Lercher, J. Am. Chem. Soc. 136 (2014) 10287-10298. DOI:10.1021/ja501592y |
| [49] |
Z. Wei, Y. Chen, J. Wang, et al., ACS Catal. 6 (2016) 5816-5822. DOI:10.1021/acscatal.6b01240 |
| [50] |
L. Zhu, Y. Jiang, J. Zheng, et al., Small 11 (2015) 4385-4393. DOI:10.1002/smll.v11.34 |
| [51] |
L. Zhu, Z. Yang, J. Zheng, et al., J. Mater. Chem. A 3 (2015) 11716-11719. DOI:10.1039/C5TA02452H |
| [52] |
N.C. Nelson, J.S. Manzano, A.D. Sadow, S.H. Overbury, I.I. Slowing, ACS Catal. 5 (2015) 2051-2061. |