 2018, Vol. 29
2018, Vol. 29 



b Toyota Research Institute-North America, Ann Arbor, MI 48105, United States;
c Department of Chemistry, University of Wisconsin-Madison, Madison, WI 53706, United States
Photocatalytic splitting of H2O to H2 and O2 has been identified as a possible green route to produce alternative, sustainable noncarbon fuel, and potential improvements to the efficiency of this process have motivated the study of this photocatalytic reaction. Titania-based photocatalysts are the most studied heterogeneous photocatalysts, being intensively investigated for water splitting and oxidation of pollutants in water and building surfaces [1-9]. Titania-based materials, however, are semiconductors with wide bandgap energies (3.0 eV for rutile and 3.2 eV for anatase) and are inefficient photocatalysts for H2 production compared to other water splitting photocatalysts [2, 3, 5, 7, 10]. Furthermore, TiO2 does not efficiently utilize the solar spectrum as activation requires excitation in the limited UV region (< 400 nm). Modification of TiO2 photocatalysts improves efficiency by broadening light absorption into the visible range (> 400 nm) or preventing recombination of photoexcited electron (e-) and hole (h+) pairs [11-16]. These improvements focus only on photoelectronic steps of the water splitting mechanism and neglect potential issues concerning the surface mechanism, such as stability of surface reaction intermediates and product desorption.
Outside of surface science studies with well-defined TiO2 single crystals [17], fundamental spectroscopic information about surface reaction intermediates and mechanisms over more typical titania powders under representative photocatalytic reaction conditions is scarce. Spectroscopic investigation of TiO2 powders during water splitting reaction conditions can provide new fundamental insights into surface reaction intermediates, mechanism, and the rate-determining-step. A primary difficulty in spectroscopic detection of surface reaction intermediate structures (i.e., hydroxyl, superoxo and peroxo species) is that their spectral vibrations [18-21] are overlapped by the strong characteristic Fourier transform infrared (FTIR) absorption bands of reactant H2O bathing the titania photocatalyst [22].
The above difficulty can be overcome through application of attenuated total reflectance (ATR)-FTIR spectroscopy, which can probe solid-liquid interfaces in heterogeneous photocatalytic systems [22-26]. Almeida et al. applied in situ ATR-FTIR to investigate cyclohexane photo-oxidation on TiO2 (anatase) photocatalysts to identify key surface reaction intermediates [27-30]. Nakamura et al. employed ATR-FTIR during photo-oxidation of water by TiO2 (rutile) powder and observed accumulation of surface hydro-peroxo Ti-OOH (838 cm-1) and peroxo Ti-OO-Ti (812 cm-1) species with time [19]. The effect of pH, however, especially in the presence of acidifying Fe3+ ions as electron scavengers, calls into question the relevance of these intermediates under pH neutral conditions [20]. The current study investigates the time-resolved surface reaction intermediates and gaseous evolution reaction rates during photocatalytic splitting of water by TiO2 powders with in situ ATR-FTIR spectroscopy under more representative reaction conditions without additives.
Overall water splitting requires the formation of H2 and O2 in a 2:1 ratio. Production of H2 and lack of O2 evolution by an aqueous slurry of TiO2 (Degussa P-25) during water splitting conditions in a UV-irradiated photo-reactor (450 W Hg lamp, > 290 nm) is shown in Fig. 1. In addition to complete lack of O2 evolution over a 5 h irradiation period, the total amount of H2 produced slows with time. The slowing production is quantified by plotting the H2 evolution rate as function of time (Fig. 1), which monotonically decreases with irradiation time. Such findings agree with previous reports for TiO2 water splitting [3] and reflect the deactivation of the TiO2 photocatalyst. The deactivation mechanism is suggested to be related to photo-reduction of O2 and subsequent reformation of H2O under dark conditions [3], but this mechanism is unconfirmed since direct in situ spectroscopic evidence has not been provided.

|
Download:
|
| Fig. 1. Total H2 production (black), total O2 production (red), and the rate of H2 production (blue) as a function of time in the photo-reactor for TiO2 with UV irradiation. | |
{kind=link}
If deactivation occurs by photo-reduction of O2, accumulation of photo-reduced surface oxygen species is expected on TiO2 during photocatalytic water splitting. Previous spectroscopic observations of accumulated surface Ti-OOH species may be an indication that TiO2 was not actually evolving O2 and, furthermore, was deactivating for H2 evolution [19]. Unfortunately, quantitative O2 and H2 evolution results were not reported therein, and furthermore, the hydroperoxo (Ti—OOH), peroxo (Ti—OO—Ti or Ti (O2)2-) or superperoxo (Ti(O2)-) intermediates were only observed at low pH in the presence of the aqueous Fe3+ ions. The spectroscopy herein will avoid the use of Fe3 when identifying stable surface intermediates during water splitting to better provide insight into the deactivation mechanism at neutral pH.
Vibrations of surface intermediates formed by adsorption of HOOH(aq) on TiO2 powder were initially measured to aid spectroscopic assignment. The time-resolved in situ ATR-FTIR spectral range shown in Fig. 2a is centered at peroxo vibrations (750–950 cm-1). Immediate appearance of the band at 877 cm-1 is assigned to the peroxo stretching νs(O-O) vibration of unbound HOOH(aq) [19, 20]. As HOOH(aq) adsorbs on TiO2 over time, a band centered at 834 cm-1 is detected, increases in intensity, and stabilizes after ~15 min. This band is assigned to νs(O—O) of adsorbed Ti—OOH hydroperoxo species [19]. These in situ ATRFTIR spectroscopic measurements identified the vibrational band for the surface Ti—OOH intermediate in a condensed water phase and agree with previous findings for the same model experimental system [19], thus the current experimental in situ ATR-FTIR system established its ability to detect accumulated Ti-OOH in the 750– 950 cm-1spectral region.

|
Download:
|
| Fig. 2. Time resolved in situ ATR-FTIR spectra centered around the (a) peroxide spectral region and (b) the superoxide spectral region during HOOH(aq) adsorption on a thin TiO2 film from a 30% HOOH/H2O solution at ambient conditions. The TiO2 in H2O spectrum was used as background. | |
{kind=link}
An additional weak ATR-FTIR band is detected at 1121 cm-1 in Fig. 2b that was not reported in previous in situ ATR-FTIR spectra during HOOH(aq) adsorption. The FTIR vibration at 1121 cm-1 is assigned to νs(O—O) stretch of adsorbed Ti(O2)- superoxide species and is consistent with superoxide νs(O—O) stretches found on reduced TiO2 (anatase), which appear in the 1060–1180 cm-1 interval depending on the degree of reduction [31].
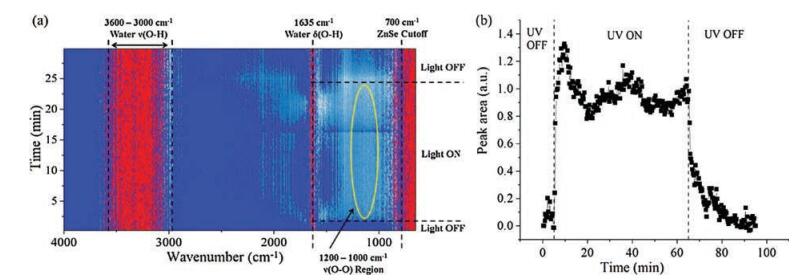
Time-resolved in situ ATR-FTIR spectra during photocatalytic splitting of H2O by TiO2 are presented as a 2D contour map in Fig. 3a (12s per spectrum). Dark blue, light blue, and red indicate areas of low, intermediate, and high intensity, respectively. Irradiation was achieved using a 150 W Xe arc lamp with broad spectrum irradiation, containing UV light from ~250–400 nm (Fig. S1 in Supporting information). The area of high intensity (red color) at ≤ 700 cm-1 is due to strong absorption of the ZnSe single crystal ATR IRE. Additional regions of high intensity are due to strong vibrational contributions of condensed H2O phase bathing the TiO2, which gives rise to vs(O-H) stretching vibrations (~3000– 3600 cm-1) and the δ(O—H) O—H scissors-bending mode (1635 cm-1) [22]. A complete discussion of the effect of water and TiO2 film thickness on resolution and the background in in situ ATR-FTIR is found in Figs. S2 and S3 in Supporting information.

|
Download:
|
| Fig. 3. (a) Time resolved 2D contour map of in situ ATR-FTIR spectra during photocatalytic water splitting on a TiO2 thin film and under dark and irradiated conditions. (b) Integrated in situ ATR-FTIR intensity of the 1000–1200 cm-1 region as a function of time for 60 min illumination. The in situ ATR-FTIR spectra were background subtracted with the ATR-FTIR signal from the condensed H2O/TiO2 system under dark conditions. | |
{kind=link}
The contour map (Fig. 3a) displays a region of increased intensity upon UV irradiation, as indicated by appearance of a light blue/white region from 1000–1200 cm-1. When UV irradiation ceased, intensity in this region decreased. The 750–1000 cm-1 region was previously predicted to contain vibrations for surface hydroperoxo Ti—OOH or peroxo Ti—OO—Ti intermediates. There is little to no increase in intensity in this region upon UV irradiation, especially compared to the change observed from 1000–1200 cm-1. In fact, the 750–1000 cm-1 region of Fig. 3a shows significant "white" intensity even in the absence of UV irradiation. Contributions at 750–1000 cm-1 in the absence of UV irradiation may be related to water penetrating the TiO2 thin film since pure water exhibits a vibrational mode below 1000 cm-1 [18]. The absence increased intensity with time at 834 cm-1, as was observed during HOOH(aq) adsorption (Fig. 2), indicates that stable Ti—OOH does not accumulate during photocatalytic water splitting conditions at neutral pH. Furthermore, the previous assignment of a shoulder at 812 cm-1 for the νs(O—O) of Ti—OO—Ti is not responsible for the increased intensity as such a feature was only found for alkaline pH [19].
Interpretation of the 1000–1200 cm-1 region is made more obvious by observation of the integrated signal (Fig. 3b), which reveals increased intensity immediately after UV irradiation begins and decreased intensity once UV irradiation ends. It has been previously reported that such a broad absorption can occur over much of the FTIR spectrum (1000–4000 cm-1) upon irradiation of TiO2, and the intensity of that absorption increases with decreasing wavenumber [32]. This "tilting" of the TiO2 FTIR spectrum is attributed to photogenerated electrons in the conduction band and/or shallow traps below the conduction band. Absorption of this nature can be used to quantify the lifetime of photo-generated electrons [32], however, appearance and decay of the broad feature is typically on far different time scales. Whereas, in the current study, appearance and decay of features from 1000–1200 cm-1 occurs on the order of minutes (Fig. 3b), appearance of broad absorption from 1000–4000 cm-1 was shown to occur almost instantaneously and decay was on the order of microseconds. Chen et al. did, however, identify a similar broad feature (1100– 1400 cm-1) that, to the current authors' knowledge, remains unassigned [32].
The FTIR vibrations of many surface dioxygen and oxygen species are found in the spectral region of 1000–1200 cm-1, especially those related to photo-reduced O2, as predicted by DFT calculations on TiO2 (anatase) (101) by Mattioli et al. [20], and a detailed discussion of the assignment is given in Supporting information. The primary surface species identified in this region are the δ(O—H) vibration of a charged hydroperoxo Ti—OOH- at 1151 cm-1, the νs(O—O) stretch for adsorbed superoxo Ti(O2)- at 1194 cm-1, and the δ(O-H) vibration for two-fold coordinated (bridged) surface Ti-(OH+)-Ti groups at 1075 cm-1. The broad nature of the experimentally detected bands is also in agreement with previously observed and unidentified broad bands in the 1000–1200 cm-1 range [18, 32], and is the broadness is attributed to the diversity of accumulated surface oxygenated species that possess vibrations in this region.
An additional experiment was performed on the TiO2 photocatalyst thin film under UV irradiated conditions, but in the absence of the liquid phase H2O reactant. In such experiment, the TiO2 photocatalyst was also exposed to molecular O2 in air. In the absence of water and exposure to molecular O2 in air (Fig. S4 in Supporting information), the increase in the 1000–1200 cm-1 region was not observed during UV irradiation of the TiO2 powder, confirming that the observations are indeed related to formation of surface oxygenated species during photocatalytic splitting of condensed water.
The accumulation of surface dioxygen and oxygen species observed above is identified as the barrier to O2 production, and the cause of deactivation of H2 production for photocatalytic water splitting over TiO2 (Fig. 1). Density functional theory calculations by Valdés et al. concluded that the rate-determining-step for photocatalytic water oxidation is photoactivation of H2O to form surface hydroxyl groups at coordinatively unsaturated TiO2 (rutile) sites and that the overpotential for O2 evolution is available under the experimental conditions of irradiation alone, i.e., no externally applied overpotential [21]. Although the photoactivation of H2O on TiO2 is the probable rate-determining-step for H2 evolution, for O2 evolution, the spectroscopic observability of surface Ti-OOH- species in the 1000–1200 cm-1 vibrational range indicates that dissociative adsorption by photoactivation of H2O is not ratedetermining for O2 production. This is concluded because additional intermediates should not accumulate after the ratedetermining-step and would, therefore, not be spectroscopically relevant. Rather, stable formation of Ti-OOH- species indicates this intermediate is involved in the rate-determining-step, e.g., the formation of adsorbed molecular O2 from Ti-OOH- or desorption of molecular O2 from TiO2.
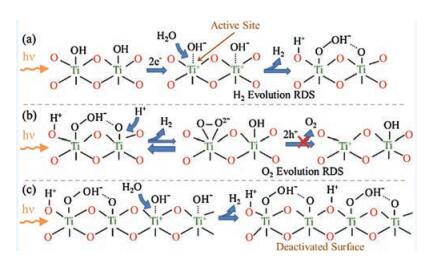
The proposed reaction mechanism for evolution of H2 from TiO2 during UV-activated photocatalytic water splitting on TiO2 is schematically shown in Fig. 4a. Spectroscopic justification of these conclusions regarding kinetics and mechanism of water splitting on TiO2 is further discussed in the Supporting information. Based on the DFT results from Valdés et al., the active site for photocatalytic water splitting is the coordinatively unsaturated Ti surface site [21]. Such sites are only applicable at low pH, whereas at neutral pH, the TiO2 is largely hydroxyl terminated. Therefore, the relevant active site for TiO2 in contact with pH neutral water, is Ti site with photoactivated terminal hydroxyl species. Dissociative adsorption of photoactivated H2O occurs at the active site and results in the formation of species observed in the 1000–1200 cm-1 FTIR region: charged Ti-OOH- and two-fold coordinated (bridged) surface Ti-(OH+)-Ti. Evolution of H2 occurs via this step and agrees with the predicted rate-determining-step for H2 evolution [21].

|
Download:
|
| Fig. 4. Schematic representation of the surface photoreaction intermediates present on TiO2 in contact with water under UV irradiation during (a) the initial production of H2, (b) proposed rate-determining-step for O2 evolution, and (c) the deactivation of H2 evolution by accumulation of oxygen surface intermediates. Note that O2 production does not occur on the TiO2 surface. | |
{kind=link}
Evolution of O2 requires dehydrogenation of stable charged Ti-OOH- to form a surface superoxo Ti(O2)- or peroxo Ti(O2)2-, that through interactions with photo-generated holes, desorbs to form O2. The broad nature of the FTIR feature between 1000 and 1200 cm-1 suggests formation of surface Ti(O2)- superoxides is possible, yet O2 evolution does not occur (Fig. 1). Peroxo Ti(O2)2- also potentially forms, however, its presence is not favored in aqueous conditions; and when formed, it is likely to become charged Ti-OOH- rather than desorb to form product O2 [20]. Thus, dehydrogenation of charged surface Ti-OOH- is the rate-determining-step for O2 evolution (Fig. 4b). Note that the bond formed between the oxygen of the neighboring Ti site and hydrogen in the Ti-OOH- is also predicted to contribute to the broad nature of the 1000–1200 cm-1 vibrational region of the ATR-FTIR spectra [20]. Once again, stability the stability of species in the FTIR supports the experimental inability to evolve O2. Hydrogen is produced as the Ti-OOH- species accumulates, but the rate of H2 production diminishes as accumulation increases and sites are blocked by stable surface Ti-OOH- species (Fig. 1). The TiO2 surface is completely deactivated when Ti-OOH- species occupy all Ti active sites (Fig. 4c).
Time-resolved in situ ATR-FTIR was employed to directly monitor surface oxygenated reaction intermediates present on TiO2 during photocatalytic water splitting. During UV irradiated photocatalytic splitting of water by TiO2 powder, multiple surface dioxygen and oxygen species are present on TiO2 (primarily charged Ti-OOH- and bridging Ti-(OH+)-Ti groups). The accumulation of such stable surface oxygenated species on TiO2 during photocatalytic splitting of water blocks surface sites required for photoactivation of H2O and is responsible for the absence of O2 evolution and decreasing H2 evolution rate with reaction time. This study is among the first to provide direct supporting evidence for the accumulation of surface oxygenated species and their blocking of available photoactive sites accounting for the poor performance of TiO2 under pH neutral conditions as an overall water splitting photocatalyst.
AcknowledgmentsWe thank Prof. Kazunari Domen and Naoyuki Sakamoto of the University of Tokyo for assistance obtaining photo-reactor data, Prof. Guido Mul and Ana Rita Almeida of TU Delft for aid in design of ATR-FTIR experiments, Jeff Christenson at Harrick Scientific for the ATR cell, and Pisist Kumnorkaew of Lehigh University for providing the thin film preparation method.
| [1] |
A. Fujishima, K. Honda, Nature 238 (1972) 37-38. DOI:10.1038/238037a0 |
| [2] |
K. Sayama, H. Arakawa, Faraday Trans. 93 (1997) 1647-1654. DOI:10.1039/a607662i |
| [3] |
K. Maeda, Chem. Commun. 49 (2013) 8404-8406. DOI:10.1039/c3cc44151b |
| [4] |
M. Anpo, M. Takeuchi, J. Catal. 216 (2003) 505-516. DOI:10.1016/S0021-9517(02)00104-5 |
| [5] |
F.E. Osterloh, Chem. Mater. 20 (2008) 35-54. DOI:10.1021/cm7024203 |
| [6] |
J. Tang, J.R. Durrant, D.R. Klug, J. Am. Chem. Soc. 130 (2008) 13885-13891. DOI:10.1021/ja8034637 |
| [7] |
A. Kudo, Y. Miseki, Chem. Soc. Rev. 38 (2009) 253-278. DOI:10.1039/B800489G |
| [8] |
J.M. Herrmann, Catal. Today 53 (1999) 115-129. DOI:10.1016/S0920-5861(99)00107-8 |
| [9] |
A. Fujishima, X. Zhang, C. R. Chim. 9 (2006) 750-760. DOI:10.1016/j.crci.2005.02.055 |
| [10] |
T. Takata, A. Tanaka, M. Hara, J.N. Kondo, K. Domen, Catal. Today 44 (1998) 17-26. DOI:10.1016/S0920-5861(98)00170-9 |
| [11] |
M.I. Litter, Appl. Catal. B 23 (1999) 89-114. DOI:10.1016/S0926-3373(99)00069-7 |
| [12] |
H. Kato, K. Asakura, A. Kudo, J. Am. Chem. Soc. 125 (2003) 3082-3089. DOI:10.1021/ja027751g |
| [13] |
K. Maeda, K. Teramura, N. Saito, Y. Inoue, K. Domen, J. Catal. 243 (2006) 303-308. DOI:10.1016/j.jcat.2006.07.023 |
| [14] |
S. Usseglio, A. Damin, D. Scarano, et al., J. Am. Chem. Soc. 129 (2007) 2822-2828. DOI:10.1021/ja066083m |
| [15] |
J. Shi, J. Chen, Z. Feng, et al., J. Phys. Chem. C 111 (2007) 693-699. DOI:10.1021/jp065744z |
| [16] |
D.B. Ingram, S. Linic, J. Am. Chem. Soc. 133 (2011) 5202-5205. DOI:10.1021/ja200086g |
| [17] |
M.A. Henderson, A Surf. Sci. Rep. 66 (2011) 185-297. DOI:10.1016/j.surfrep.2011.01.001 |
| [18] |
R. Nakamura, A. Imanishi, K. Murakoshi, Y. Nakaoto, J. Am. Chem. Soc. 125 (2003) 7443-7450. DOI:10.1021/ja029503q |
| [19] |
R. Nakamura, Y. Nakato, J. Am. Chem. Soc. 126 (2004) 1290-1298. DOI:10.1021/ja0388764 |
| [20] |
G. Mattioli, F. Filippone, A.A. Bonapasta, J. Am. Chem. Soc. 128 (2006) 13772-13780. DOI:10.1021/ja062145x |
| [21] |
A. Valdés, Z.W. Qu, G.J. Kroes, J. Rossmeisl, J.K. Nørskov, J. Phys. Chem. C 112 (2008) 9872-9879. DOI:10.1021/jp711929d |
| [22] |
B.L. Mojet, S.D. Ebbeson, L. Lefferts, Chem. Soc. Rev. 39 (2010) 3643-4655. |
| [23] |
A.R. Hind, S.K. Bhargava, A. McKinnon, Adv. Coll. Int. Sci. 93 (2001) 91-114. DOI:10.1016/S0001-8686(00)00079-8 |
| [24] |
T. Bürgi, A. Baiker, J. Phys. Chem. B 106 (2002) 10649-10658. DOI:10.1021/jp0255987 |
| [25] |
I. Ortiz-Hernandez, C.T. Williams, Langmuir 19 (2003) 2956-2962. DOI:10.1021/la020799n |
| [26] |
J.M. Andanson, A. Baiker, Chem. Soc. Rev. 39 (2010) 4571-4584. DOI:10.1039/b919544k |
| [27] |
A.R. Almeida, J.A. Moulijn, G. Mul, J. Phys. Chem. C 112 (2008) 1552-1561. |
| [28] |
A.R. Almeida, J.T. Carneiro, J.A. Moulijn, G. Mul, J. Catal. 273 (2010) 116-124. DOI:10.1016/j.jcat.2010.05.006 |
| [29] |
A.R. Almeida, R. Berger, J.A. Moulijn, G. Mul, Phys. Chem. Chem. Phys. 13 (2011) 1345-1355. DOI:10.1039/C0CP00879F |
| [30] |
A.R. Almeida, J.A. Moulijn, G. Mul, J. Phys. Chem. C 115 (2011) 1330-1338. DOI:10.1021/jp107290r |
| [31] |
A. Davydov, Molecular Spectroscopy of Oxide Catalyst Surfaces[M]. Hoboken: John Wiley & Sons, 2003.
|
| [32] |
T. Chen, Z. Feng, G. Wu, et al., J. Phys. Chem. C 111 (2007) 8005-8014. DOI:10.1021/jp071022b |