 2018, Vol. 29
2018, Vol. 29 



Titanium dioxide (TiO2) has various practical applications in many important fields, such as semiconductor electrodes in photochemical cells, coating materials used for UV protection, photocatalysts for water splitting, sensing and photochromic materials, etc. [1, 2]. It has been intensively studied by wide range of experimental and theoretical methods [3-5]. However, for its different polymorphs, anatase TiO2 is relatively less investigated compared with rutile [10-13], though it usually exhibits higher activities in catalysis. As the most stable and abundant facets exposed at these two TiO2 nanomaterials, anatase TiO2(101) and rutile TiO2(110) have been carefully studied. In particular, several probe molecules, such as NO, O2, CO, HCOOH, have been chosen to study the surface properties of these facets. In fact, such probe molecules are also important in practice, as O2 is one of the most common gases in the air and the main product in photocatalytic water splitting, and NO is also one of the main components in automotive exhaust [6-9].
A number of theoretical studies on the adsorptions of NO and O2 at rutile TiO2(110) are available [10-13], while those on anatase TiO2(101) are rather limited. Nevertheless, it has been found that defect sites on these surfaces, including step edges, surface hydroxyls, oxygen vacancies and interstitial titanium [14-17], can modify their structural and electronic properties and act as the active sites for various applications. In particular, the surface hydroxyls attracted great attention since they are popular surface species at TiO2 in real use and related to surface processes in photocatalysis. Wendt et al. [18] and Liu et al. [19] found that the hydroxylated rutile TiO2(110) can significantly affect the diffusion of H2O and the adsorptions of O2. The presence of surface hydroxyl is also essential for the covalent coupling of aryl halide monomers on the rutile TiO2(011) [20]. In addition, Liu et al. [21] showed that the hydroxyls on rutile surfaces can greatly promote the transformation of NO2 into HNO3.
In order to further explore the catalytic behaviors of surface hydroxyls at anatase TiO2, we calculated the adsorption properties of NO and O2 at hydrogenated anatase TiO2(101). Various surface adsorption sites were considered in the calculations, and it has been determined that the hydrogenation can affect the surface electronic structures and increase the adsorption strengths of the NO and O2 molecules through charge transfer. Interestingly, we also found that, different from the hydrogenated rutile TiO2(110), the hydrogenated anatase TiO2(101) can favor the adsorptions at restricted positions beside the hydroxyl, and further analyses indicate that the local bonding configurations mainly contribute to such unique properties.
In this work, all the calculations were conducted using the Vienna ab initio simulation package (VASP) [22, 23], and the electron exchange-correlation was described by the PBE-GGA functional [24]. The core-valence electron interaction was represented by the project-augmented wave (PAW) [25, 26] method. The calculations also included the on-site Coulomb corrections [27, 28] (DFT + U, U = 4.2 eV for Ti 3d states as suggested by Deskins et al. [29] and Yoon et al. [30]) and long-range dispersion interactions [31, 32] (DFT-D). The kinetic energy cut-off of 400 eV was used for the plane wave basis-set expansion. The titanium (3s, 3p, 3d, 4s), the hydrogen (1s), and the nitrogen and oxygen (2s, 2p) electrons were treated as valence electrons.
The anatase TiO2(101) surface was modeled as a periodic slab with four TiO2 layers of oxide, and the vacuum between slabs was ~10 Å. The 2 × 3 surface cell and corresponding 2 × 2 ×1 k-point mesh were used in the calculations. The adsorption was modeled on one side of the slab, and during structural optimizations, all of the atoms except those in the bottom TiO2 layer of the slab were allowed to relax until the atomic forces reached below 0.05 eV/Å.
To estimate the adsorption energies (Eads) of the molecules (NO and O2), the following expression was considered:
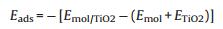
|
where Emol/TiO2 is the total energy of the system with one adsorbed molecule and TiO2 surface slab; ETiO2 is the total energy of the surface slab; and Emol is the total energy of a single molecule in gasphase.
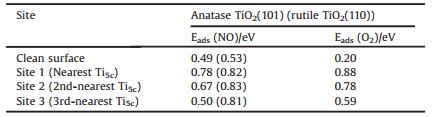
The well-known structures of stoichiometric anatase TiO2(101) and rutile TiO2(110) are illustrated in Fig. 1. Both fully saturated 3 (6)-fold coordinated O(Ti) (O3c and Ti6c) atoms and coordinatively unsaturated 2(5)-fold coordinated O(Ti) (O2c and Ti5c) are exposed on these surfaces.

|
Download:
|
| Fig. 1. Calculated structures (side view) of the stoichiometric (a) anatase TiO2(101) and (b) rutile TiO2(110). Ti and O atoms are in light grey and red, respectively. | |
It is generally believed that the stoichiometric terminations of these most stable facets of anatase and rutile TiO2 are relatively unreactive. At the same time, it has been found that the occurrence of defective species would increase their activities. Yu et al. [33] reported that on the stoichiometric rutile TiO2(110), the adsorption energy of NO is as low as 0.53 eV, and it can be significantly increased to 0.82 eV when the surface is hydrogenated. Hu and coworkers [19] determined that the adsorption energy of O2 would increase by 0.5 eV on the hydrogenated rutile TiO2(110). It also needs to be mentioned that, according to these studies, the various Ti5c sites beside the surface O2cH hydroxyl (Fig. 1b) give almost no obvious difference regarding the adsorption energies of NO and O2.
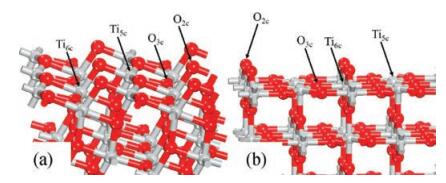
In the current work, we also chose the clean and hydrogenated anatase TiO2(101) to calculate and compare their reactivities. As one can see from Fig. 2a, the exposed Ti5c binds with four O3c and one O2c; the Ti5c-O3c bonds are slightly above 2.0 Å, while the Ti5cO2c bond is as short as 1.86 Å. Following hydrogenation, the O2cH hydroxyl occurs and the original Ti5c-O2c bond is significantly stretched to as long as 2.07 Å though the other bonds are only slightly stretched.

|
Download:
|
| Fig. 2. Calculated structures (top view) of the (a) stoichiometric and (b) hydrogenated anatase TiO2(101), and (c) NO and (d) O2 being adsorbed on stoichiometric anatase TiO2(101) (the insets are corresponding side views). Several important calculated bond lengths are also given. Three sites (Site 1, Site 2 and Site 3) on the hydrogenated surface where NO and O2 adsorptions were calculated are specified in (b). O of adsorbed molecules are in pink, and N in blue. | |
Then, we calculated the adsorptions of NO and O2 on the clean anatase TiO2(101) surface firstly. The most favorable adsorption site for the molecules is again Ti5c (Fig. 2), and the calculated Eads are 0.49 eV and 0.20 eV for NO and O2, respectively. On the hydrogenated anatase TiO2(101), we then considered the adsorption Ti5c sites at different positions with respect to the hydroxyl (Fig. 2b). In Table 1, we list the calculated adsorption energies of these two molecules at the different Ti5c sites. As one can see from the table, the calculated adsorptions of NO and O2 at the three different positions exhibit very different performance compared to those at rutile TiO2(110). The NO and O2 molecules gave very different adsorption energies at these three different sites, and in particular, for NO, its adsorption strength is drastically increased only at Site 1, which is just beside the hydroxyl, while it is barely affected at other sites further away from the hydroxyl. Similarly, O2 adsorption is also heavily enhanced at such nearest Site 1, while the enhancement effect is less significant at Site 2 and further decreases at the Site 3.
|
|
Table 1 Calculated adsorption energies of NO and O2 at clean and different sites (Site 1–3) of hydrogenated anatase TiO2(101), with comparison of those of NO at clean and various Ti5c of hydrogenated rutile TiO2(110) (in parentheses) [33]. |
{kind=link}
{kind=link}
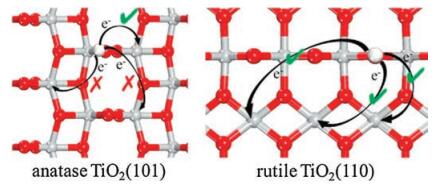
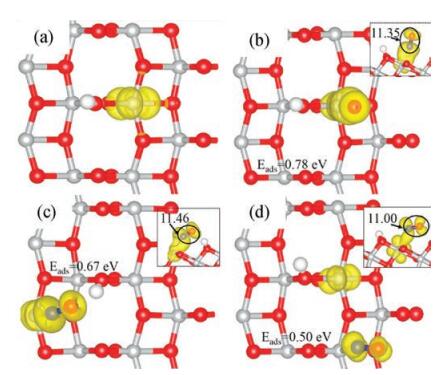
In order to better understand the different performances of NO and O2 adsorptions at rutile TiO2(110) and anatase TiO2(101), we conducted analyses from electronic and structural points of view. Yu et al. [33] reported that on the hydrogenated rutile TiO2(110), the single electron transferred from the adsorbed H could be localized at different nearby surface Ti5c (nearest, second-nearest and third-nearest to the surface hydroxyl) with similar stabilities. On the contrary, our calculations in the current work determined that the localized electron (Fig. 3a) at the hydrogenated anatase TiO2(101) can occur only at the nearest Ti5c (Fig. 2b, Site 1). It is well known that the localized electron at TiO2 surfaces can turn the Ti cation where it is localized from one Ti4+ into a Ti3+, which will then have a significantly larger ion radius. Accordingly, the local bonding structures will also be affected by the occurrence of Ti3+, i.e., the Ti-O bonds should be stretched due to the large Ti3+ ion radius. At clean and hydrogenated rutile TiO2(110), each Ti5c still binds with five O3c and therefore they each have similar capacity to take the localized electron, and such relatively weak bonds with O3c can be also readily stretched.

|
Download:
|
| Fig. 3. Calculated spin-polarized charge densities (in yellow) of (a) the hydrogenated anatase TiO2(101) and the surfaces with the adsorbed NO at (b–d) Site 1, 2 and 3. The corresponding adsorption energies are also listed, as well as the calculated Bader charges of the NO (see insert, side view). | |
{kind=link}
However, the situation is completely different at anatase TiO2(101). Besides the four O3c, the Ti5c at the clean surface now also binds with one O2c which is much more reactive than the O3c, and as we have illustrated in the above, the Ti5c-O2c bond then becomes much shorter (Fig. 2a). Consequently, the Ti5c at hydrogenated anatase TiO2(101) is less feasible to increase its radius compared to that at rutile TiO2(110), and only the one directly binding with the hydroxyl can be readily reduced since such hydrogenated O2c is now 3-fold coordinated and binding less strongly with the Ti5c (Fig. 2b).
The combined electronic and structural effects discussed in the above can not only explain the different performances of the localized electron at anatase TiO2(101) and rutile TiO2(110), and they may also help understand the adsorptions of NO and O2. In Figs. 3b-d, we plot the calculated spin-polarized charge densities of the systems with adsorbed NO at different Ti5c sites of the hydrogenated anatase TiO2(101). As one can see, for the NO at Site 1 and Site 2, the originally localized electron (Fig. 3a) is now partially transferred to the NO (see the calculated Bader charges of NO in the insert) and largely shared by it and the Ti5c below. Therefore, the adsorption at Site 2 is less favorable since the Ti5c cation is binding with a clean O2c and less likely to take the partial electron compared to the one of Site 1. For the NO adsorbed at Site 3, it gains no extra electron and the localized electron is still at the Ti5c of Site 1 as a whole. Therefore, the adsorption energy is nearly identical to that at stoichiometric anatase TiO2(101).
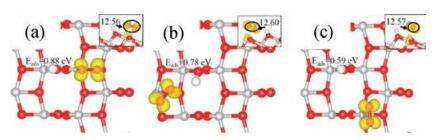
Similar electronic analyses were also conducted on the anatase TiO2(101) surfaces with adsorbed O2 at different sites (Figs. 4a-c). Different from the case with adsorbed NO, the O2 at all the three sites can take partial electron and become negatively charged, which could be due to the relatively lower 2π* orbital of O2 compared to that of NO where the partial electron stays. Again, the different stabilities of the adsorption systems with O2 can be explained by the positions of the Ti5c with the localized (partial) electron. In addition, the existence of H-bond between the hydroxyl and the adsorbed O2 may also contribute to the stronger adsorption of O2 at Site 2 compared to that at Site 3.

|
Download:
|
| Fig. 4. Calculated spin-polarized charge densities (in yellow) of the hydrogenated anatase TiO2(101) with the adsorbed O2 at (a–c) Site 1, 2 and 3. The corresponding adsorption energies are also listed, as well as the calculated Bader charges of the O2 (see insert, side view). | |
{kind=link}
In this work, we calculated the hydrogenated anatase TiO2(101) by focusing on its electronic and structural properties and activities toward adsorptions of NO and O2. We found that, different from the hydrogenated rutile TiO2(110), the hydrogenated anatase TiO2(101) can generate the localized electron at only one specific surface Ti cation that directly binds with the hydroxyl, and favor the NO and O2 adsorptions at such position. Detailed analyses indicate that the local bonding configurations mainly contribute to these unique properties. The results obtained in this work also suggest that in photocatalytic processes, anatase TiO2(101) with the excited electron at a surface Ti5c may give rise to the specifically activated O2c directly binding with such Ti3+, where unique activities such as proton adsorption and water dissociation may occur since these processes can efficiently stabilize the enlarged Ti3+.
AcknowledgmentsWe are grateful for financial support from the National Natural Science Foundation of China (Nos. 21421004, 21573067, 91545103) and Program of Shanghai Academic Research Leader (No. 17XD1401400). The authors also thank the National Super Computing Center in Jinan for computing time.
| [1] |
A. Fujishima, T.N. Rao, D.A. Tryk, J. Photochem. Photobiol. C 1 (2000) 1-21. DOI:10.1016/S1389-5567(00)00002-2 |
| [2] |
X.B. Chen, S.S. Mao, Chem. Rev. 107 (2007) 2891-2959. DOI:10.1021/cr0500535 |
| [3] |
Z.L. Hua, Z.Y. Dai, X. Bai, et al., Chem. Eng. J. 283 (2016) 514-523. DOI:10.1016/j.cej.2015.07.072 |
| [4] |
A. Yamamoto, Y. Mizuno, K. Teramura, S. Hosokawa, T. Tanaka, Appl. Catal. BEnviron. 180 (2016) 283-290. DOI:10.1016/j.apcatb.2015.06.036 |
| [5] |
R. Kaplan, B. Erjavec, G. Dražic, J. Grdadolnik, A. Pintar, Appl. Catal. B-Environ. 181 (2016) 465-474. DOI:10.1016/j.apcatb.2015.08.027 |
| [6] |
Q.J. Jin, Y.S. Shen, S.M. Zhu, J. Coll. Interface Sci. 487 (2017) 401-409. DOI:10.1016/j.jcis.2016.10.056 |
| [7] |
A. Yamamoto, K. Teramura, S. Hosokawa, T. Tanaka, Sci. Technol. Adv. Mater. 16 (2015) 024901. DOI:10.1088/1468-6996/16/2/024901 |
| [8] |
B. Shen, T. Liu, N. Zhao, X.Y. Yang, L.D. Deng, J. Environ. Sci. 9 (2010) 1447-1454. |
| [9] |
L. Qiu, Y. Wang, D.D. Pang, et al., Catalysts 6 (2016) 6010009. |
| [10] |
H.H. Li, X.Y. Wu, S. Yin, K. Katsumata, Y.H. Wang, Appl. Surf. Sci. 392 (2017) 531-539. DOI:10.1016/j.apsusc.2016.09.075 |
| [11] |
X.Y. Xie, Q. Wang, W.H. Fang, G.L. Cui, J. Phys. Chem. C 121 (2017) 16373-16380. DOI:10.1021/acs.jpcc.7b04811 |
| [12] |
S.C. Li, P. Jacobson, S.L. Zhao, X.Q. Gong, U. Diebold, J. Phys. Chem. C 116 (2012) 1887-1891. DOI:10.1021/jp209290a |
| [13] |
R. Niishiro, R. Konta, H. Kato, et al., J. Phys. Chem. C 111 (2007) 17420-17426. DOI:10.1021/jp074707k |
| [14] |
M. Fronzi, W. Daly, M. Nolan, Appl. Catal. A-Gen. 521 (2016) 240-249. DOI:10.1016/j.apcata.2015.11.038 |
| [15] |
M.F. Camellone, P.M. Kowalski, D. Marx, Phys. Rev. B 84 (2011) 2507-2524. |
| [16] |
P. Ganesh, J. Phys. Chem. Lett. 2 (2011) 2918-2924. DOI:10.1021/jz2013177 |
| [17] |
U. Martinez, L.B. Vilhelmsen, H.H. Kristoffersen, Phys. Rev. B:Condens. Matter. 84 (2011) 3239-3247. |
| [18] |
S. Wendt, J. Matthiesen, R. Schaub, et al., Phys. Rev. Lett. 96 (2006) 066107. DOI:10.1103/PhysRevLett.96.066107 |
| [19] |
L.M. Liu, B. McAllister, H.Q. Ye, P. Hu, J. Am. Chem. Soc. 128 (2006) 4017-4022. DOI:10.1021/ja056801p |
| [20] |
M. Kolmer, R. Zuzak, A.A. Ahmad Zebari, et al., Chem. Commun. (Camb.) 51 (2015) 11276-11279. DOI:10.1039/C5CC02989A |
| [21] |
C. Liu, Q.X. Ma, H. He, et al., Environ. Sci.:Nano 4 (2017) 2388-2394. DOI:10.1039/C7EN00920H |
| [22] |
G. Kresse, J. Furthmüller, Comp. Mater. Sci. 6 (1996) 15-50. DOI:10.1016/0927-0256(96)00008-0 |
| [23] |
G. Kresse, J. Furthmüller, Phys. Rev. B 54 (1996) 11169-11186. DOI:10.1103/PhysRevB.54.11169 |
| [24] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [25] |
G. Kresse, D. Joubert, Phys. Rev. B 56 (1999) 1758-1775. |
| [26] |
P.E. Blöchl, Phys. Rev. B:Condens. Matter 50 (1994) 17953-17979. |
| [27] |
S.L. Dudarev, G.A. Botton, S.Y. Savrasov, C.J. Humphreys, A.P. Sutton, Phys. Rev. B 57 (1998) 1505. DOI:10.1103/PhysRevB.57.1505 |
| [28] |
V.I. Anisimov, F. Aryasetiawan, A.I. Lichtenstein, J. Phys. Condens. Matter. 9 (1997) 767. DOI:10.1088/0953-8984/9/4/002 |
| [29] |
N.A. Deskins, R. Rousseau, M. Dupuis, J. Phys. Chem. C 115 (2011) 7562-7572. DOI:10.1021/jp2001139 |
| [30] |
Y. Yoon, Y. Du, J.C. Garcia, et al., Phys. Chem. Chem. Phys. 16 (2015) 313-321. DOI:10.1002/cphc.v16.2 |
| [31] |
S. Grimme, J. Comput. Chem. 27 (2006) 1787-1799. DOI:10.1002/(ISSN)1096-987X |
| [32] |
S. Grimme, J. Comput. Chem. 25 (2004) 1463-1473. DOI:10.1002/(ISSN)1096-987X |
| [33] |
Y.Y. Yu, U. Diebold, X.Q. Gong, Phys. Chem. Chem. Phys. 17 (2015) 26594-26598. DOI:10.1039/C5CP04584C |