 2018, Vol. 29
2018, Vol. 29 



Titanium dioxide (TiO2) is an inexpensive, non-toxic and ubiquitous material widely studied in photocatalysis for applications including water splitting [1-3], CO2 reduction [4-7] and pollutant degradation [8, 9]. While it is robust and stable under operating conditions, the large bandgap (> 3 eV) means that TiO2 is UV active and light absorption must be extended into the visible range to achieve absorption of solar energy for widespread use of photocatalysis. Modification of TiO2 to induce visible light absorption are widely studied [1-3, 8].
Significant effort has focused on substitutional cation and/or anion doping at Ti or O sites respectively [10-19]. These modifications introduce dopant-derived states into the band gap of the TiO2 host, which can facilitate electronic transitions with energies in the visible range. However, localised defect states have been shown to act as recombination centres, impeding carrier migration and reducing photocatalytic activity [17]. Another approach is co-doping of a metal and non-metal at Ti and O sites [20-26]. Passivation of defect levels through co-doping with charge compensating n-p pairs has been proposed [20, 22] and realised [25], and non-compensating co-doping schemes have also been suggested [26]. However, practical issues with reproducibility, solubility and stability can still persist with doped TiO2. Another approach is to modify the surface of TiO2 to simultaneously promote visible light absorptionwhile maintaining efficient charge separation. The development of the dye sensitised solar cell (DSSC) [27] has realised this in part, and similar strategies can be used in photocatalysis. Improved photocatalytic efficiency in the UV and visible has been reported for TiO2 films and nanopillars decorated with metal nanoparticles (surface plasmon resonance) [28-30]. The drawback here is that precious metals such as Ag, Au and Pt are used and this drives up costs.
Surface modification with dispersed metal oxide nanoclusters of non-precious metals has yielded promising results. Fabrication of such composites has been demonstrated experimentally via chemisorption-calcination cycle (CCC) [31, 32] and atomic layer deposition (ALD) [33]. These studies reported on FeOx-modified TiO2 and found both band gap reduction and enhanced visible light photocatalytic activity. This was attributed to a decrease in carrier recombination as revealed by photoluminescence spectroscopy [31]. Density functional theory (DFT) simulations corroborated Xray photoelectron spectroscopy (XPS) data in identifying cluster derived states at the valence band maximum (VBM) that shift the VBM to higher energy leading to band gap reduction [31, 32, 34]. The size of the nanocluster modifier tunes absorption through the dependence of the energy gap on nanocluster composition and the presence of low-coordinated sites in the cluster facilitates electron and hole trapping and adsorption and activation of molecules [35]. Thus, surface modification with metal oxide nanoclusters is an attractive prospect.
DFT has been used in combination with experiment to understand these systems [6, 32, 34-46]. Our previous work [6, 32, 35, 38, 39, 41-43, 46, 47] has indicated the potential for metal oxide modifiers to induce a bandgap reduction over bare TiO2. We have highlighted the importance of the TiO2 crystal form [47] through SnO2-modified rutile and anatase, the oxidation state of the supported nanocluster cation, through SnO/SnO2 and PbO/ PbO2 [43] and the role of cation-derived valance band states in SnO/PbO/Bi2O3 modified TiO2 [38-40].
Inducing visible light absorption should not be the sole focus as charge separation and chemical reactivity also play significant roles. An enhanced separation of photoexcited electrons and holes has been reported to result from nanocluster surface modification of TiO2 [35, 39, 40, 43, 48] and surface restructuring of the (001) surface of anatase TiO2 [49]. Di Valentin and Selloni studied charge localisation and separation in photoexcited anatase [50]. In this model a triplet electronic state is imposed, which promotes an electron to the conduction band (CB) with a valence band (VB) hole. We have used this model to highlight the role of low-coordinated nanocluster metal and/or oxygen sites in trapping and separating charge carriers [35, 39, 40, 43] and this suggests that modification of TiO2 promotes electron and hole separation.
In this paper, we present a DFT study of TiO2anatase (101) modified with sub-nm nanoclusters of SnO and MgO, with specific compositions Sn4O4 and Mg4O4. We focus in particular on the impact of nanocluster modification on (1) the interfacial atomic structure, (2) the valence or conduction band edges of anatase (101), (3) charge localisation after excitation and (4) the reducibility of the composite system. Point defects, such as oxygen vacancies, govern the chemistry at metal oxide surfaces by providing sites for the adsorption and activation of molecules such as H2O and CO2. Previous work highlighted the significance of vacancy formation energies in predicting the strength of interaction of CO2 molecules at the reduced cluster-TiO2 surface [6]. Importantly, we go beyond the perfect, clean surface models of TiO2 and investigate the impact of surface hydroxylation, through adsorption of dissociated water, on the nanocluster adsorption and stability, building on previous work [37, 51] which showed that surface hydroxylation has a significant impact on the properties of the nanocluster-TiO2 composite system.
We predict that modification of anatase (101) will induce a red shift in light absorption due to the emergence of nanocluster derived electronic states that shift the valence band to higher energy, irrespective of the stat of the anatase surface. Surface modification promotes charge separation, with electrons and holes localising on surface cation and nanocluster anion sites respectively. Finally, we show that the reducibility of the composite systems is enhanced over bare anatase (101) and we predict that the composite surfaces will be more reactive. Combined with a red shift in light absorption, these results yield an insight into the design of new photocatalytic materials.
All calculations were performed using periodic plane wave DFT as implemented in the VASP5.2 code [52, 53], with details given in the supplementary material. The effect of surface hydroxyl species on the interfacial chemistry is studied with a model where four water molecules are adsorbed dissociatively at anatase (101) corresponding to a 50% water coverage. To consistently describe the partially filled Ti3d states that form after electron localisation from excitation or oxygen vacancy formation, a Hubbard +U correction is applied, with a value of U(Ti) = 4.5 eV [40, 41, 43, 54]. For calculations involving excited states and valence band hole formation we applied the +U correction to the O2p state with U (O) = 5.5 eV, which localises oxygen holes [37-40, 43, 55].
The nanoclusters are adsorbed in different configurations at anatase (101) and the adsorption energies are calculated using:
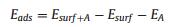
|
(1) |
Esurf + A, Esurf and EA are the energies of the adsorbate-surface composite system, the bare/hydroxylated TiO2 surface slab and the gas phase nanocluster. A negative adsorption energy indicates exothermic adsorption of the nanocluster. We model photoexcitation by imposing a triplet electronic state. This promotes an electron to the CB with a corresponding hole in the VB. We evaluate the energetics and charge localisation associated with photoexcitation, as described in the Supporting information.
We begin our analysis with the characteristics of the bare and hydroxylated anatase (101) surfaces, hence forth denoted as oxidised (o-anatase) and hydroxylated (oh-anatase). Fig. S2a (Supporting information) shows the atomic structure of the ideal, extended anatase (101) surface, free from point defects and surface hydroxyls. The surface is terminated by alternating rows of twofold (O2f) and three-fold (O3f) coordinated oxygen atoms. Of the two sublayers of Ti atoms, the outermost consists of rows of fivefold coordinated Ti5f atoms and the next sublayer has rows of 6-fold coordinated Ti6f atoms. With 16 Ti atoms accessible on our (2 × 4) surface supercell, the eight outermost Ti5f atoms act as sites for water adsorption. In our model hydroxylated surface we adsorb four dissociated water molecules as shown in Fig. S2b (Supporting information) at 50% coverage. This is not aiming for the most stable water adsorption structure at anatase (101) surface, but is a reasonable model to explore the influence of surface hydroxylation.
We adopt the following notation for the different atoms present in the composites: Surface oxygen atoms are denoted OS and further differentiated by coordination number (O2f and O3f), as are surface Ti atoms (Ti5f and Ti6f). Cluster oxygen atoms are denoted OC and oxygen atoms present in hydroxyls are labelled OW.
The adsorption energies of the nanoclusters at the o-anatase and oh-anatase (101) surfaces are -5.73 eV and -7.49 eV for Mg4O4 and -1.89 eV and -2.16 eV for Sn4O4. The negative values indicate a favourable nanocluster-surface interaction while the magnitudes suggest that the nanoclusters will be stable against desorption and surface migration. The relaxed atomic structures are presented in Fig. 1. The most stable gas phase geometries for the Mg4O4 and Sn4O4 nanoclusters are cubes and adsorption on the anatase (101) surface modifies these structures substantially. This allows for the formation of more metal-oxygen interfacial bonds which strengthens the interaction between the cluster and the surface.

|
Download:
|
| Fig. 1. Atomic structures of Mg4O4-o-anatase (a, b), Mg4O4-oh-anatase (101) (c, d), Sn4O4-o-anatase (e, f), Sn4O4-oh-anatase (101) (g, h). Insets of panels on the left show adsorption energies. Atoms of the clusters are enlarged for clarity. Ti is light grey, O is red, H is white, Mg is green and Sn is dark grey spheres. | |
For Mg4O4, the interaction between the nanocluster and both surfaces is strong with more favourable adsorption at the hydroxylated surface. The atomic structure of Mg4O4 adsorbed at the o-anatase (101) surface is shown in Figs. 1a and b. Three of the four cluster oxygen atoms form a bond with surface Ti5f atoms. Two Mg atoms in the cluster make two bonds with surface O2f and O3f atoms and are four-fold coordinate. The other Mg atoms are three-fold coordinate with one sharing a bond with a single surface O2f and the other bonded only to OC atoms. Adsorption involves the formation of eight bonds between the nanocluster and the bare surface; of these three are Ti-OC bonds with lengths in the range of 1.9 -2.1 Å. The Ti atoms concerned migrate out from the Ti5f plane by up to 0.4 Å. Four of the five interfacial Mg-OS bonds have distances in the range of 2.1-2.2 Å; the fifth Mg-OS distance, which involves a three-fold coordinated Mg cation, is 1.9 Å. The single O3f site involved migrates out from the surface layer by 0.3 Å; this distortion breaks the bond between the O3f site and the Ti6f subsurface atom.
Comparing with adsorption at the oh-anatase (101) surface (Figs. 1c and d) we see that only two of the OC atoms form bonds with surface Ti5f atoms with Ti-O distances of 1.9 Å and 2.0 Å. This is to be expected as the presence of the OH groups results in fewer Ti5f atoms being accessible. However, one Mg atom forms bonds with four surface oxygens (two O2f and two O3f) to make it six-fold coordinated, with Mg-OS distances of 2.1-2.2 Å. Another Mg atom is four-fold coordinated and binds with two surface O2f and an OW atom from an adsorbed OHT group. The two remaining cluster cations are three-fold coordinated with one sharing a bond with an OW and the other bonded only to cluster oxygens. The MgO-anatase interaction at the oh-anatase surface results in surface Ti atoms that form bonds to the nanocluster migrating out of the Ti5f plane to a more significant degree than observed in adsorption at the o-anatase surface. In one instance in which a surface Ti atom shares bonds with both the supported nanocluster and an adsorbed OHT group the surface cation migrates out from the Ti5f plane by 1.0 Å and this distortion leads to the breaking of two Ti-OS bonds. The two O3f atoms that form bonds to the nanocluster migrate out from the surface layer by 0.4 Å.
Turning now to the Sn4O4 nanocluster, the interaction of Sn4O4 with the o-anatase and oh-anatase (101) surfaces is weaker than that of the Mg4O4 nanocluster. Nanocluster adsorption is more favourable at the hydroxylated surface, by 0.27 eV. We can attribute the weak adsorption to the formation of fewer interfacial bonds between SnO and anatase compared to MgO and anatase, with only five and four interfacial bonds formed for adsorption at the bare and hydroxylated surfaces respectively. Figs. 1e and f show the atomic structure of the Sn4O4 nanocluster adsorbed at the oanatase (101) surface. Two oxygen atoms in the nanocluster do not bind with the anatase surface. The third oxygen forms a bond with a single surface Ti5f cation and the fourth OC atom is two-fold coordinated sharing one interfacial bond with a surface Ti5f cation. This contrasts with the Mg4O4 case in which each of the cluster OC atoms are three-fold coordinated with three forming bonds with the surface. Each of the four Sn atoms in the nanocluster are threefold coordinated and three have interfacial bonds with a single row of bridging surface O2f atoms. The two Ti-OC distances are 1.8 Å and 1.9 Å, with the shorter distance corresponding to the lowercoordinated OC site. The Sn-OS distances are in the range of 2.1 - 2.3 Å. For adsorption of the Sn4O4 nanocluster at the oh-anatase (101) surface (Figs. 1g and h), none of the cluster oxygen atoms bind to the surface and all four interfacial bonds involve three-fold coordinated cluster Sn atoms. Two of these bind to OW atoms, with Sn-OW distances of 2.1 Å and 2.3 Å, and two to bridging surface O2f atoms with bond lengths of 2.1 Å and 2.2 Å.
Upon adsorption and relaxation of both nanoclusters at the ohanatase (101) surface we observe migration of H atoms from surface hydroxyls to bind with OC atoms. In the Mg4O4 case, one H atom migrates from a surface O2f hydroxyl (OHB) to a two-fold coordinated OC atom. This facilitates the formation of new bonds between a Mg site and the surface O2f site. Another H atom migrates from an adsorbed OHT group to bind with a nanocluster oxygen leaving a terminal singly coordinated OW bound to a surface Ti5f. This is accompanied by a distortion in which the surface Ti is drawn out from the Ti5f plane by 0.6 Å, breaking a bond with a subsurface OS leaving the oxygen atom two-fold coordinated. Migration of H atoms from the hydroxylated surface to oxygen sites of the adsorbed Mg4O4 nanocluster makes each OC site threefold coordinated as in the Mg4O4-o-anatase (101) case.
When the Sn4O4 nanocluster adsorbs at the oh-anatase surface two H atoms migrate from surface O2f atoms to OC atoms. Again this facilitates the formation of interfacial Sn-O bonds through the provision of more low-coordinated OS sites to which the Sn cations can bind. However, in this instance not all OC sites are rendered three-fold coordinated through this migration; one OC site is twofold coordinated such that the coordination configuration of the Sn4O4 nanocluster at the hydroxylated surface is the same as that at the bare surface.
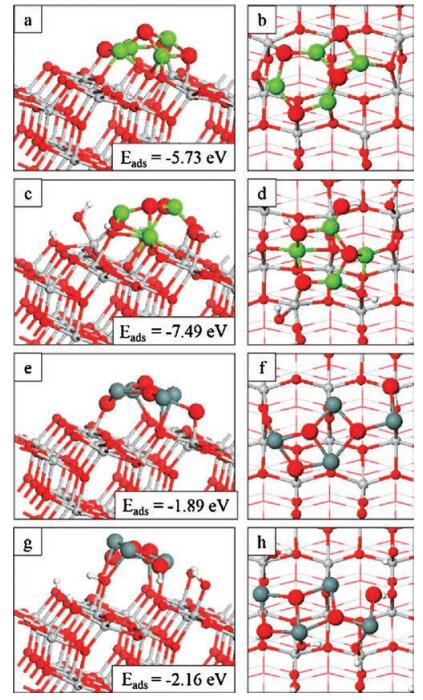
Figs. 2a and b display the calculated spin polarised projected electronic density of states (PEDOS) of the Mg4O4 nanocluster modifying the o-anatase and oh-anatase (101) surfaces respectively. In each instance the top panels show the contribution to the density of states (DOS) due to surface Ti 3d and cluster Mg 3s and 3p states. The bottom panels show the contribution due to O 2p states, separated according to whether the oxygen atom is found in the surface (OS), the supported nanocluster (OC) or adsorbed water/hydroxyl (OW).

|
Download:
|
| Fig. 2. Spin polarized projected electron density of states plots for (a) Mg4O4-o-anatase (101), (b) Mg4O4-oh-anatase (101), (c) Sn4O4-o-anatase (101) and (d) Sn4O4-ohanatase (101). Top panels display metal PEDOS and bottom panels display oxygen 2p PEDOS. Inset in (b) shows overlapping contributions from OC and OW 2p states. | |
Examination of the PEDOS shows that modification of o-anatase and oh-anatase (101) with Mg4O4 introduces new states into the gap just above the VB edge. Focusing on Mg4O4 at the o-anatase (101) surface (Fig. 2a) we see that modification results in an enhanced DOS at the VB edge and the emergence of OC 2p derived states around 0.5eV above the VBM. From this qualitative description of the DOS we predict a red shift in the band gap due to the Mg4O4 modification, which could lead to improved photoactivity in the visible range. Enhanced UV activity is also postulated due to the increased DOS at the VB edge and the presence of empty Mg derived states above the CB edge [56].
Similar features are observed in the PEDOS of Mg4O4 modifying the oh-anatase (101) surface (Fig. 2b). However, the presence of hydroxyl species on the surface in combination with the Mg4O4 modification appears to extend the valence band to higher energy, into the anatase bandgap, pushing the VB edge to 0.5eV above that of bare TiO2 as shown in the inset of Fig. 2b. The overall effect on the bandgap is comparable to that observed in the bare surface and the same conclusions may be drawn regarding improvements in activity in both the visible and UV range.
The top panels of Figs. 2c and d present the Ti3d and Sn 5s and 5p PEDOS for Sn4O4 modifying the o-anatase (101) and oh-anatase (101)surfaces respectively; bottompanels show DOS contributions from OS, OC and OW 2p. In both instances features emerge in the anatase energy gap which are attributable to the surface modification. For Sn4O4 modifying the o-anatase (101) surface (Fig. 2c) the impact on the electronic band structure is similar to that of the Mg4O4 modification (Fig. 2a). OC 2p derived states are present at the top of the TiO2-derived VB and contribute to a new state that lies 0.5eV above the TiO2 VBM. Sn-derived states also appear in the TiO2 bandgap at around 0.5eV above the VBM with empty Sn-derived states lying above the TiO2 CB edge. This arises from the Sn2+ oxidation state and the highest lying occupied states show contributions from both Sn 5s/5p and O 2p states [40, 57, 58].
The effect of modification on the electronic structure is more dramatic for Sn4O4 atthe oh-anatase(101) surface (Fig. 2d). OW and OC 2p states enhance the DOS at the TiO2 VB edge with some OC 2p derived states appearing as much as 1eV above the VBM. Sn-5s/5p derived states lie at similar positions at the top of the VB.
For each of the four heterostructures presented we propose that modification will result in a reduction of the energy gap of anatase TiO2, thus inducing a red shift and extending light absorption into the visible range. We also predict improved UV photocatalytic activity with the alignments of the surface and nanocluster energy bands facilitating charge separation.
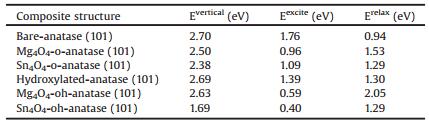
Table 1 presents the energies computed from the model of the photoexcited state, discussed in the methodology. The underestimation of the bandgap inherent in approximate DFT is present in the current DFT+U computational set-up. The +U corrections are chosen to localise electrons and holes rather than to reproduce the bandgap of bulk TiO2. This underestimation is clear in the computed values for Evertical and Eexcite which are clearly smaller than the experimental values. However, what is important is the change in these quantities with modification of the anatase surfaces. The excess spin density plots and PEDOS plots for the unmodified bare and hydroxylated anatase (101) surfaces can be found in Figs. S3 and S4 (Supporting information).
|
|
Table 1 Vertical singlet-tripletenergy difference (Evertical), the relaxed singlet-tripletenergy difference (Eexcite) and the relaxation energy (Erelax) for nanocluster modified oanatase and oh-anatase (101). Values for unmodified o-anatase and oh-anatase (101) have been included for reference. |
{kind=link}
{kind=link}
We note that Eexcite is always smaller than the simple VB-CB energy gap and Evertical, as the former energy includes the ionic relaxations and polaron formation in response to "exciting" the electron which then lowers the energy of the triplet electronic state. However, comparison of these computed energies across different structures yields valid qualitative information [35, 38-40, 43, 47]. In particular, a reduction in Eexcite for a composite structure relative tothe unmodified metal oxide will correspond to light absorption at lower energies for the surface modified system.
Table 1 shows that the modification of anatase, whether bare or hydroxylated, with MgO and SnO nanoclusters always results in a red shift, with both the vertical and excitation energies being reduced upon modification. The reduction in the excitation energy arises from the greater degree of structural relaxation in the nanocluster, as evidenced by the values for Erelax. The relaxation, or trapping energy, is an indication of the stability of the electronhole pair and the larger relaxation energy for hydroxylated anatase indicates a higher stability of the localised electron-hole pair compared to the o-anatase (101) surface.
Looking at differences between the nanoclusters, we see that MgO modification has a more significant effect than SnO modification of the o-anatase (101) surface; modification with Mg4O4 and Sn4O4 gives reductions in Eexcite of 1.80eV and 0.67eV relative to the bare surface. The opposite is true for the oh-anatase (101) surface; Eexcite is calculated to be lower by 0.80 (0.99) eV upon modification with Mg4O4 (Sn4O4). These results, in combination with analysis of the ground state PEDOS, indicate a red shift in light absorption for the modified surfaces. In terms of the stability of the localised electrons and holes, Mg4O4 modification shows significantly enhanced electron and hole trapping at both the bare and hydroxylated surfaces. The presence of hydroxyls at the surface facilitates the trapping of the electron-hole pair for both the bare surface and that modified with Mg4O4 but has no impact on the trapping energy of the Sn4O4-modified surface.
In addition to energy gap considerations, another factor affecting the efficiency of photocatalytic materials is the fate of photoexcited charge carriers. Charge recombination must be suppressed and charge separation promoted. In addition, a higher relaxation energy indicates higher stability of the photo induced charges. We have examined the localisation of photoexcited charges through analysis of computed Bader charges, spin magnetisations and excess spin density plots.
Figs. 3a-d show the spin density plots for the Mg4O4-modified o-anatase and oh-anatase (101) surfaces after relaxation of the triplet state. For both modified surfaces we see that the electron localises on a single surface Ti5f atom. The computed Bader charge for Ti4+ cations is 1.27 electrons (net Bader charge of +2.73 electrons). The Bader charge increases to 1.70 electrons (net charge of +2.3 electrons) at the site on which the electron is localised. These Bader charges are consistent with formation of a localised Ti3+ electronic state. The computed spin magnetisation for the Ti3+ site is 0.99 μB and compares with values of less than 0.03 μB for all other Ti sites. Finally, the Ti-O distances around this site are elongated by 0.1 Å relative to the singlet ground state which is typical of the formation of a localised, reduced Ti3+ state.

|
Download:
|
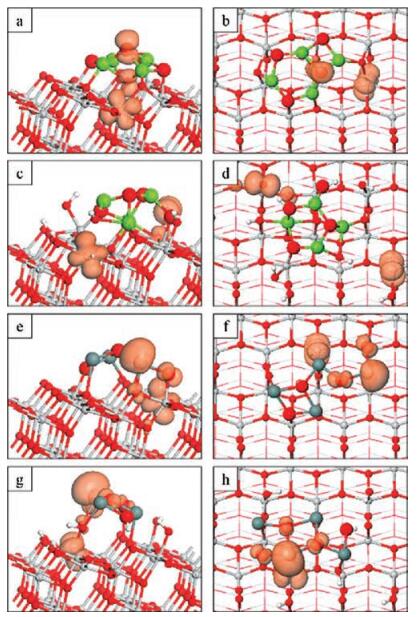
| Fig. 3. Spin density plots for the photoexcited electron and hole in Mg4O4-oanatase (a, b), Mg4O4-oh-anatase (101) (c, d), Sn4O4-o-anatase (e, f), Sn4O4-ohanatase (101) (g, h). The orange spin density isosurfaces enclose spin densities up to 0.2 eV/Å3. | |
{kind=link}
For Mg4O4 supported on the bare surface (Figs. 3a and b) the hole state predominantly localises at a three-fold coordinated oxygen site in the nanocluster. The computed Bader charge for the OC atom is reduced from 7.93 electrons to 7.23 electrons (net charges of -1.93 and -1.23 electrons) upon localisation of the hole state. The computed spin magnetisation is 0.67μB, which is usual for a localised oxygen hole in the DFT + U formalism. These values indicate the formation of a localised oxygen hole state. There is some spreading of the hole state to a neighbouring surface O2f site which shares multiple bonds with the nanocluster. This spreading is reflected in a computed spin magnetisation of 0.21 μB at the O2f site and an elongation of the Ti6f-O2f bond by 0.1 Å and the breaking of the Ti5f-O2f bond.
For Mg4O4 supported on the oh-anatase (101) surface (Figs. 3c and d) we can see that the hole state localises at a singly coordinated, originally hydroxyl oxygen site, OW. Hole localisation is accompanied by a reduction in the Bader charge for the terminal oxygen from 7.16 electrons (net charge of -1.16 electrons) in the singlet state to 6.73 electrons (net charge of -0.73 electrons) in the triplet state. The computed spin magnetisation is 0.81 μB. Again these values are typical of a localised oxygen hole species. The Ti-O distance is elongated by 0.2 Å relative to that in the singlet ground state, consistent with the formation of a localised oxygen holepolaron.
The spin density plots for the Sn4O4-modified o-anatase (Figs. 3e and f) and oh-anatase (101) (Figs. 3g and h) surfaces show an electron localised on a single Ti lattice site. In both instances, these are surface Ti5f and the computed Bader charges (spin magnetisations) are 1.72 (0.97) electrons (μB) and 1.67 (0.99) electrons (μB) on the o-anatase and oh-anatase (101) surfaces respectively. These results indicate the formation of a reduced, localised Ti3+ species and further confirmation is provided by the distortions of the atomic structure in the vicinity of the localisation. The Ti3+-O bond lengths increased by as much as 0.13 Å in the excited state relative to the equivalent distances in the ground state.
For Sn4O4 modifying the o-anatase (101) surface (Figs. 3e and f) the hole state is distributed over two doubly-coordinated OC atoms and a cluster Sn atom to which they are each bonded. This localisation arises because the top of the valence band is composed of Sn 5s + 5p and O 2p states from the SnO nanocluster. The computed Bader charges decrease from 7.3-7.4 and 12.64 electrons for oxygen and tin in the singlet ground state, to 7.15-7.18 and 12.36 electrons for the same atoms in the relaxed triplet state. Considering that a Bader charge of 12.75 electrons is computed for Sn2+ ions in the gas phase Sn4O4 nanocluster, we assign a 3+ oxidation state to the Sn atom at which hole localisation occurs. Correspondingly a-1 oxidation state (oxygen hole) is assigned to the two OC sites over which the hole is distributed. The charge is uniformly spread over the three sites with spin magnetisations of 0.19, 0.22 and 0.24μB computed for the two oxygen sites and the tin site respectively. These values compare with computed spin magnetisations of less than 0.01 μB on the other Sn and O sites; there is a small distribution of charge to a neighbouring OS site as shown in Fig. 3e and this is reflected in a computed spin magnetisation of 0.06 μB. Electron localisation is accompanied by an extension of the Ti-OC bond by 0.1 Å but the distances between the various sites over which the hole is distributed are contracted relative to the ground state. The two Sn3+-OC and the Sn3+-OS bond lengths are reduced by 0.09-0.15 Å and this distortion contributes to the breaking of a Sn-OC bond involving one of the oxygen polaron sites; the OC in question was three-fold coordinated in the ground state (Fig. 1f) and is now two-fold coordinated in the excited state (Fig. 3f) so that both OC sites at which hole localisation occurs are two-fold coordinated.
For the Sn4O4-modified oh-anatase (101) surface (Figs. 3g and h), we see that the hole state is distributed over a tin site and three oxygen sites in the supported Sn4O4 cluster. The computed Bader charge and spin magnetisations are 12.26 electrons and 0.28 μB for the Sn site, to which we assign a +3 oxidation state. The computed spin magnetisations for the three OC sites are 0.08, 0.15 and 0.15 μB; the computed Bader charges are 7.25, 7.62 and 7.65 electrons. The distances between the cluster sites over which the hole state is distributed are contracted with a reduction of 0.09- 0.17 Å in the Sn3+-OC bond lengths.
For all systems studied, the model photoexcited state results in the localisation of an electron at a surface Ti5f site to form a reduced Ti3+ polaron species. This is consistent with the nature of the lowest lying conduction band states, which are dominated by Ti 3d states. The nature of the hole states depends on the identity of the nanocluster modifier and the state of the anatase (101) surface. For modification with the Mg4O4 nanocluster, the hole can localise at a single OC site (bare surface) or OW site (hydroxylated surface). For the Sn4O4 nanocluster modifier, the hole is distributed over metal and oxygen sites in the nanocluster.
The proximity of the charge carriers to each other contrasts with previous work on surface modified rutile (110) where electron localisation occurred at subsurface Ti sites [35, 38, 43]. Still, the stability of the electron and hole and their spatial separation are improved relative to the bare anatase (101) surface (Fig. S3 in Supporting information). Modification with larger nanoclusters may improve charge separation, as the general trend observed in previous work is that hole localisation occurs at the lowercoordinated sites in the cluster [35, 38-40]. However, the impact of an increase in cluster size on the light absorption properties of the heterostructures would need to be reassessed due to well-known size effects.
Surface modification promotes separation of photoexcited charge, with holes, localised on or distributed over sites of the nanocluster, available for participation in oxidation reactions. However, for this separation and localisation to result in enhanced photocatalytic activity, reductant species must preferentially adsorb at the nanocluster modifier rather than the TiO2 support. The investigation of the adsorption of feedstock species, such as H2O or CO2, at various sites of the modified surfaces is the subject of ongoing work.
Labelling the cluster oxygen atoms Ⅰ-Ⅳ (Fig. S5 in Supporting information), we examine oxygen vacancy formation by removing each nanocluster oxygen and relaxing. The oxygen vacancy formation energy is computed from:

|
(2) |
where Esurf, and Esurf+vac are the computed total energies for surface modified anatase (101) before and after vacancy formation respectively, and EO2 is the energy of a free gas phase O2 molecule. The results of these calculations, presented in Table S1 of Supporting information, allow us to examine the reducibility of modified anatase and any impact of surface hydroxylation. The relaxed geometries of the composite surfaces upon formation of the most stable oxygen vacancy are presented in Fig. 4.

|
Download:
|
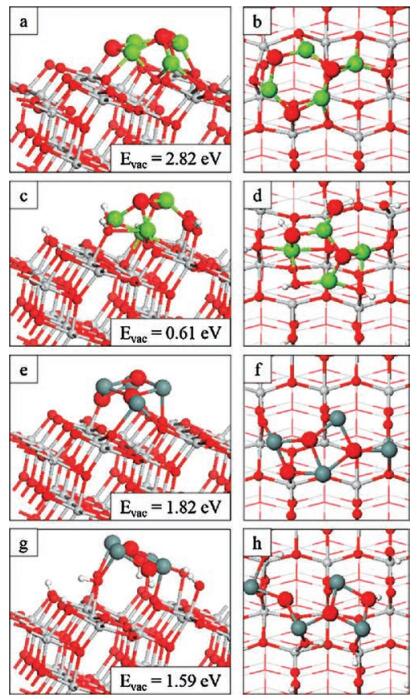
| Fig. 4. Relaxed atomic structure of the nanocluster modified TiO2 surfaces after formation of the most stable oxygen vacancies. (a, b) Mg4O4-o-anatase (101), (c, d) Mg4O4-oh-anatase (101), (e, f) Sn4O4-o-anatase (101) and (g, h) Sn4O4-oh-anatase (101). Insets of panels on the left show vacancy formation energies. | |
{kind=link}
For MgO-o-anatase (101), the most stable oxygen vacancy has a formation energy of 2.82 eV, while for SnO-o-anatase (101), the cost is 1.82 eV. Hence, the energy required to produce the most stable oxygen vacancy in the nanocluster-modified surface is notably reduced over the unmodified anatase surface for which the formation energy of an oxygen vacancy is 3.6-3.8 eV from a study with similar computational parameters [59]. For the MgO-o-anatase (101) system, the most favourable vacancy site is OⅢ and the relatively high energy cost is probably due to the resulting under-coordination of the cluster metal ions in the nonstoichiometric structure as a result of a lack of oxygen sites with which the Mg atoms can form bonds. This is, however, the most stable site as it was the only site for which all cluster metal ions were at least three-fold coordinated upon relaxation as shown in Figs. 4a and b. However, the oxygen vacancy necessitated the formation of additional interfacial Mg-O3f bonds. This draws the O3f atoms out from the surface, breaking Ti6f-O3f bonds and leaving Ti6f ions under-coordinated at the surface. Thus the surface distortions that accompany oxygen vacancy formation act against the gain in energy when the structure relaxes in response to oxygen vacancy formation.
For the Sn4O4 modifier at the o-anatase (101) surface (Figs. 4e and f), the most stable oxygen vacancy site in the nanocluster has a cost of 1.8 eV. The high energy cost to form the other vacancies are a result of the under-coordination of the cluster Sn ions in the resulting non-stoichiometric structures. For all sites, except the most stable, at least one Sn atom takes two-fold coordination upon vacancy formation. For the most stable oxygen vacancy the removed oxygen atom was originally two-fold coordinated. The single Sn site which was bound to the removed oxygen atom maintains its three-fold coordination by forming a new bond with a surface O3f site; this means that the atoms of the nanocluster maintain the same coordination configuration as in the stoichiometric case. The O3f site migrates out from the surface by 0.3 Å to accommodate this bond and this breaks a bond with a subsurface Ti site. The Ti5f site to which the removed oxygen was bound relaxes back into the surface and rebinds to an OS site in the substrate to remain five-fold coordinated. Turning now to the hydroxylated, modified surface, the problem of cation undercoordination that results in a large vacancy formation energy for Mg4O4-anatase is alleviated when the nanocluster is supported on the hydroxylated surface. This is because the surface terminating OH groups provide additional sites, close to the nanocluster, for the formation of new interfacial metal-oxygen bonds that maintain the Mg coordination. Two of the four Mg cations are four-fold coordinated, strengthening the interaction with the surface (Figs. 4c and d). This is reflected in quite favourable oxygen vacancy formation energies of 0.67 eV and 0.61 eV for two of the four nanocluster oxygen sites. The Mg-O interaction is strong enough that a surface Ti5f site is rendered fourfold coordinated upon reduction, with an OHT group migrating from the Ti site to a Mg site in the nanocluster.
For SnO-modified oh-anatase, (Figs. 4g and h) each of the OC and Sn atoms are three-fold coordinated. The reduced Sn4O3 nanocluster interacts too weakly to form the interfacial bonds necessary to stabilise it at the surface; however, in the presence of hydroxyls at the surface, the energy required to produce the most stable oxygen vacancy, at 1.59 eV, is reduced by 0.2 eV relative to the Sn4O4-modified o-anatase (101) surface. The formation of a neutral oxygen vacancy releases two electrons. Through an analysis of the excess spin density plots (Figs. S10 and S11 in Supporting information) and computed Bader charges and spin magnetisations we find that, for all systems, the electrons localise at two surface Ti sites. Electrons localise at low-coordinated Ti sites, whether these are Ti5f sites or Ti6f sites which have lower coordination due to surface restructuring in response to nanocluster adsorption and reduction. For Mg4O3 at the hydroxylated surface, one electron localises at a four-fold coordinated Ti site. The exception is Sn4O3 at the hydroxylated surface where electron localisation occurs at two Ti5f sites which are six-fold coordinated due to the presence of OHT species; these OHTspecies form Sn-OW bonds with the reduced nanocluster. In all instances, electron localisation is accompanied by an increase in the computed Bader charges of 0.4 electrons; spin magnetisations are in the range of 0.97-1.00 μB. These values indicate the formation of reduced Ti3+ species. It is difficult to predict a priori the impact of surface hydroxylation on oxygen vacancy formation. For the Mg4O4 modifier, the formation energy decreased in the presence of surface hydroxylation; while this was true of the Sn4O4 modifier, the effect was much less pronounced. We expect that these results depend on the geometry of the nanoclusters and the degree of surface coverage with hydroxyl groups. The strength of interaction at the nanocluster-surface interface may also play a role, particularly the metal-surface interaction.
In conclusion, we have modified oxidised and hydroxylated anatase (101) surfaces with MgO and SnO nanoclusters. Our results show that surface modification enhances the DOS at the VBM, to give rise to a red shift in light absorption. Improved charge separation is shown in a photoexcited model, in which electrons localise at surface Ti atoms and holes localise at low-coordinated nanocluster or hydroxyl sites. The presence of surface hydroxyls improves nanocluster adsorption by facilitating formation of interfacial bonds. Hydroxyl O 2p states contribute to the DOS at the VBM and a red shift is predicted. The presence of surface hydroxyls increases the stability of the photogenerate delectronhole pair. The reduction of the composite is significantly enhanced compared to bare anatase (101), so that this modification will result in a more reactive surface. The role played by surface hydroxyls in the reducibility of the modified surfaces depends on nanocluster composition. For Mg4O4-anatase surface hydroxyls lead to a significant decrease in the energy cost to reduce the composite. These results show that surface modification with MgO and SnO can impart properties important for photocatalytic applications. The DFT analysis in the present paper can inform the design and aid in the screening of new photocatalytic materials.
AcknowledgmentsWe acknowledge support from Science Foundation Ireland through the US-Ireland R & D Partnership Program (No. SFI 14/US/ E2915) and the European Commission through COST Action CM1104 "Reducible Metal Oxides, Structure and Function". We acknowledge computing resources at Tyndall funded by SFI and by the SFI and Higher Education Authority funded Irish Centre for High End Computing.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2017.11.036.
| [1] |
V. Etacheri, C. Di Valentin, J. Schneider, D. Bahnemann, S.C. Pillai, J. Photochem. Photobiol. C:Photochem. Rev. 25 (2015) 1-29. DOI:10.1016/j.jphotochemrev.2015.08.003 |
| [2] |
A. Fujishima, X. Zhang, D.A. Tryk, Surf. Sci. Rep. 63 (2008) 515-582. DOI:10.1016/j.surfrep.2008.10.001 |
| [3] |
M. Ni, M.K.H. Leung, D.Y.C. Leung, K. Sumathy, Renew Sustain. Energy Rev. 11 (2007) 401-425. DOI:10.1016/j.rser.2005.01.009 |
| [4] |
X. Chang, T. Wang, J. Gong, Energy Environ. Sci. 9 (2016) 2177-2196. DOI:10.1039/C6EE00383D |
| [5] |
N.M. Dimitrijevic, B.K. Vijayan, O.G. Poluektov, et al., J. Am. Chem. Soc. 133 (2011) 3964-3971. DOI:10.1021/ja108791u |
| [6] |
M. Fronzi, W. Daly, M. Nolan, Appl. Catal. A:Gen. 521 (2016) 240-249. DOI:10.1016/j.apcata.2015.11.038 |
| [7] |
S.C. Roy, O.K. Varghese, M. Paulose, C.A. Grimes, ACS Nano 4 (2010) 1259-1278. DOI:10.1021/nn9015423 |
| [8] |
M. Pelaez, N.T. Nolan, S.C. Pillai, et al., Appl. Catal. B:Environ. 125 (2012) 331-349. DOI:10.1016/j.apcatb.2012.05.036 |
| [9] |
S.C. Pillai, U.L. Štangar, J.A. Byrne, A. Pérez-Larios, D.D. Dionysiou, Chem. Eng. J. 261 (2015) 1-2. DOI:10.1016/j.cej.2014.11.001 |
| [10] |
C. Di Valentin, E. Finazzi, G. Pacchioni, et al., Chem. Phys. 339 (2007) 44-56. DOI:10.1016/j.chemphys.2007.07.020 |
| [11] |
X. Nie, S. Zhuo, G. Maeng, K. Sohlberg, Int. J. Photoenergy 2009 (2009) 22. |
| [12] |
K. Yang, Y. Dai, B. Huang, M.H. Whangbo, J. Phys. Chem. C 113 (2009) 2624-2629. |
| [13] |
J. Yu, Q. Xiang, M. Zhou, Appl. Catal. B:Environ. 90 (2009) 595-602. DOI:10.1016/j.apcatb.2009.04.021 |
| [14] |
J.W. Zheng, A. Bhattcahrayya, P. Wu, et al., J. Phys. Chem. C 114 (2010) 7063-7069. |
| [15] |
A.M. Czoska, S. Livraghi, M. Chiesa, et al., J. Phys. Chem. C 112 (2008) 8951-8956. DOI:10.1021/jp8004184 |
| [16] |
H. Peng, J. Li, S.S. Li, J.B. Xia, J. Phys.:Condens. Matter 20 (2008) 125207. DOI:10.1088/0953-8984/20/12/125207 |
| [17] |
J.M. Herrmann, New J. Chem. 36 (2012) 883-890. DOI:10.1039/c2nj20914d |
| [18] |
T. Ikeda, T. Nomoto, K. Eda, et al., J. Phys. Chem. C 112 (2008) 1167-1173. DOI:10.1021/jp0752264 |
| [19] |
J.P. Xu, L. Li, L.Y. Lv, et al., Chin. Phys. Lett. 26 (2009) 097502. DOI:10.1088/0256-307X/26/9/097502 |
| [20] |
S. Na Phattalung, S. Limpijumnong, J. Yu, Appl. Catal. B:Environ. 200 (2017) 1-9. DOI:10.1016/j.apcatb.2016.06.054 |
| [21] |
C.D. Valentin, G. Pacchioni, H. Onishi, A. Kudo, Chem. Phys. Lett. 469 (2009) 166-171. DOI:10.1016/j.cplett.2008.12.086 |
| [22] |
Y. Gai, J. Li, S.S. Li, J.B. Xia, S.H. Wei, Phys. Rev. Lett. 102 (2009) 036402. DOI:10.1103/PhysRevLett.102.036402 |
| [23] |
R. Long, N.J. English, J. Phys. Chem. C 114 (2010) 11984-11990. |
| [24] |
R. Long, N.J. English, Chem. Mater. 22 (2010) 1616-1623. DOI:10.1021/cm903688z |
| [25] |
J. Zhang, C. Pan, P. Fang, J. Wei, R. Xiong, ACS Appl. Mater. Interfaces 2 (2010) 1173-1176. DOI:10.1021/am100011c |
| [26] |
W. Zhu, X. Qiu, V. Iancu, et al., Phys. Rev. Lett. 103 (2009) 226401. DOI:10.1103/PhysRevLett.103.226401 |
| [27] |
M. Grätzel, J. Photochem. Photobiol. C:Photochem. Rev. 4 (2003) 145-153. DOI:10.1016/S1389-5567(03)00026-1 |
| [28] |
W. Hou, S.B. Cronin, Adv. Funct. Mater. 23 (2013) 1612-1619. DOI:10.1002/adfm.v23.13 |
| [29] |
M. Honda, Y. Kumamoto, A. Taguchi, Y. Saito, S. Kawata, Appl. Phys. Lett. 104 (2014) 061108. |
| [30] |
S. Shuang, R. Lv, Z. Xie, Z. Zhang, Sci. Rep. 6 (2016). |
| [31] |
Q. Jin, M. Fujishima, H. Tada, J. Phys. Chem. C 115 (2011) 6478-6483. DOI:10.1021/jp201131t |
| [32] |
H. Tada, Q. Jin, A. Iwaszuk, M. Nolan, J. Phys. Chem. C 118 (2014) 12077-12086. DOI:10.1021/jp412312m |
| [33] |
J.A. Libera, J.W. Elam, N.F. Sather, T. Rajh, N.M. Dimitrijevic, Chem. Mater. 22 (2010) 409-413. DOI:10.1021/cm902825c |
| [34] |
M. Nolan, Phys. Chem. Chem. Phys. 13 (2011) 18194-18199. DOI:10.1039/c1cp21418g |
| [35] |
M. Nolan, A. Iwaszuk, K.A. Gray, J. Phys. Chem. C 118 (2014) 27890-27900. DOI:10.1021/jp508822v |
| [36] |
M. Nolan, A. Iwaszuk, A.K. Lucid, J.J. Carey, M. Fronzi, Adv. Mater. 28 (2016) 5425-5446. DOI:10.1002/adma.v28.27 |
| [37] |
M. Fronzi, A. Iwaszuk, A. Lucid, M. Nolan, J. Phys.:Condens. Matter 28 (2016) 074006. DOI:10.1088/0953-8984/28/7/074006 |
| [38] |
A. Lucid, A. Iwaszuk, M. Nolan, Mater. Sci. Semicond. Process. 25 (2014) 59-67. DOI:10.1016/j.mssp.2014.01.005 |
| [39] |
A. Iwaszuk, M. Nolan, Catal. Sci. Technol. 3 (2013) 2000-2008. DOI:10.1039/c3cy00194f |
| [40] |
A. Iwaszuk, M. Nolan, J. Mater. Chem. A 1 (2013) 6670-6677. DOI:10.1039/c3ta10647k |
| [41] |
A. Iwaszuk, P.A. Mulheran, M. Nolan, J. Mater. Chem. A 1 (2013) 2515-2525. DOI:10.1039/c2ta01582j |
| [42] |
A. Iwaszuk, M. Nolan, Q. Jin, M. Fujishima, H. Tada, J. Phys. Chem. C 117 (2013) 2709-2718. DOI:10.1021/jp306793r |
| [43] |
M. Nolan, ACS Appl. Mater. Interfaces 4 (2012) 5863-5871. DOI:10.1021/am301516c |
| [44] |
M. Nolan, Chem. Commun. 47 (2011) 8617-8619. DOI:10.1039/c1cc13243a |
| [45] |
J.B. Park, J. Graciani, J. Evans, et al., J. Am. Chem. Soc. 132 (2009) 356-363. |
| [46] |
A. Iwaszuk, M. Nolan, Phys. Chem. Chem. Phys. 13 (2011) 4963-4973. DOI:10.1039/c0cp02030c |
| [47] |
Q. Jin, M. Fujishima, M. Nolan, A. Iwaszuk, H. Tada, J. Phys. Chem. C 116 (2012) 12621-12626. |
| [48] |
S.J.A. Moniz, S.A. Shevlin, X. An, Z.X. Guo, J. Tang, J. Chem Eur. 20 (2014) 15571-15579. DOI:10.1002/chem.v20.47 |
| [49] |
F. Xiong, L.L. Yin, Z. Wang, et al., J. Phys. Chem. C 121 (2017) 9991-9999. DOI:10.1021/acs.jpcc.7b02154 |
| [50] |
C. Di Valentin, A. Selloni, J. Phys. Chem. Lett. 2 (2011) 2223-2228. DOI:10.1021/jz2009874 |
| [51] |
K.C. Schwartzenberg, J.W.J. Hamilton, A.K. Lucid, et al., Catal. Today 280 (2017) 65-73. DOI:10.1016/j.cattod.2016.06.002 |
| [52] |
G. Kresse, D. Joubert, Phys. Rev. B 59 (1999) 1758-1775. |
| [53] |
J. Furthmüller, J. Hafner, G. Kresse, Phys. Rev. B 53 (1996) 7334-7351. DOI:10.1103/PhysRevB.53.7334 |
| [54] |
B.J. Morgan, G.W. Watson, Surf. Sci. 601 (2007) 5034-5041. DOI:10.1016/j.susc.2007.08.025 |
| [55] |
A. Iwaszuk, M. Nolan, J. Phys.:Condens. Matter 23 (2011) 334207. DOI:10.1088/0953-8984/23/33/334207 |
| [56] |
M. Nolan, A. Iwaszuk, H. Tada, Aust. J. Chem. 65 (2012) 624-632. DOI:10.1071/CH11451 |
| [57] |
A. Walsh, G.W. Watson, J. Phys. Chem. B 109 (2005) 18868-18875. DOI:10.1021/jp051822r |
| [58] |
A. Walsh, G.W. Watson, Phys. Rev. B 70 (2004) 235114. DOI:10.1103/PhysRevB.70.235114 |
| [59] |
M.A. Haa, A.N. Alexandrova, J. Chem. Theory Comput. 12 (2016) 2889-2895. DOI:10.1021/acs.jctc.6b00095 |