 2018, Vol. 29
2018, Vol. 29 



b Graduate School of EEWS, Korea Advanced Institute of Science and Technology(KAIST), Daejeon 305-701, Republic of Korea
Many industrial chemical processes associated with the synthesis of chemicals, environmental applications, and the production of clean energy are based on heterogeneous catalysis [1-4]. Therefore, studying the nature of heterogeneous catalysis has been an important practical task for decades. Diverse technologies have been developed to manufacture advanced catalysts that combine materials (e.g., metals, alloys, semiconductors, and oxides) with various properties, sizes, and shapes (e.g., nanoparticles, fine powders, porous materials) [1, 2, 5-7]. It is now possible to obtain catalysts with activities that are orders of magnitude greater than the activity of single-chemical catalysts. Nevertheless, even in the presence of such advanced catalysts, industrial chemical processes still occur at temperatures well above ambient temperatures. This makes the chemical industry one of the most energy-intensive technologies of our time, which motivates scientists around the world to search for new, more energy-efficient solutions in the field of heterogeneous catalysis.
One of the key technologies for increasing the activity and selectivity of heterogeneous catalysts is the use of catalytically active metal nanoparticles in combination with a suitable support [2, 8-12]. This technology is based on groundbreaking research by Schwab et al., who in the 1960s discovered significant changes in the activity of fine metal particles when placed on the support of a metal oxide, even though the support itself is inactive for this reaction [13, 14]. Called the "Schwab effect", this phenomenon is attributed to the formation of a Schottky barrier at the metal-oxide interface with the subsequent transfer of charge carriers through the barrier, which affects the course of the surface reaction [1, 9, 13]. Later, it was also shown by Somorjai et al. that additional charge transfer through a metal-support interface can occur during exothermic surface reactions through the nonadiabatic creation of excited (hot) charge carriers in the metal [15-17]. It is expected that the creation of hot charge carriers can significantly affect the rate of reagent molecule adsorption on the catalytic surface and thus influence the overall rate of the reaction [1, 5, 18-21]. Although many studies have been performed to clarify the role of hot charge carriers in catalysis, a complete understanding of this phenomenon is still lacking because of the complexity of the quantitative description of interfacial charge transfer under reaction conditions.
The nature of hot charge carriers created in metal catalysts during nonadiabatic surface reactions is analogous to that of highly excited electron-hole (e-h) pairs generated by incident light absorption in semiconductor-based photocatalysts and plasmonic photocatalysts containing nanoparticles of noble metals [19, 22-27]. However, the mechanism for the effect of hot charge carriers on the rate of chemical reactions is different. In photocatalysts, excited charge carriers are created by an influx of energy from external sources (e.g., the sun). After excitation, these charge carriers can transmit energy to molecules adsorbed on the catalytic surface and break old or form new chemical bonds, thereby converting photon energy into chemical energy. In exothermic reactions, the chemical transformations themselves are a source of energy sufficient for the generation of highly excited e-h pairs. In this case, the transfer of hot charge carriers only affects the rate of energy exchange between the metal catalyst and the support without introducing additional energy into the system. Thus, the creation of hot charge carriers from nonadiabatic energy dissipation is a process inverse to the generation of e-h pairs in photocatalysts.
This review considers the main aspects of research aimed at developing methods for studying the transfer of charge carriers through metal-support interfaces under catalytic reaction conditions. First, we review the mechanisms for hot electron excitation and transport through metal-oxide interfaces. We then show various schemes for detecting hot electrons that are generated during catalytic processes. In the end, we provide an overview of the latest results from the detection of hot electrons in supported catalysts during chemical reactions at both the gas/solid and liquid/solid interfaces and discuss the relationship between charge flow and reaction kinetics.
2. Principle of hot electron generationThe creation of hot charge carriers during gas-surface interactions has been known for a long time because of the observation of photon and electron exoemission from the highly exothermic reactions of alkali metals with various gases (e.g., Cl2, N2O, and O2) [28-38]. It is also known that the excitation of e-h pairs leads to a hot electron with energy above the Fermi level and to a hot hole with energy below the Fermi level. The excitation of an e-h pair takes place via a nonadiabatic process [2].
One view to justify the generation of hot electrons is related to the difference in heat capacity between electrons and phonons. Hot electrons thermalize within picoseconds by electron-phonon interactions. The phenomenon of hot electron creation can be rationalized as follows: The electronic heat capacity (Celectron) of most metals in thermal equilibrium is about one hundred times smaller than the lattice heat capacity (Clattice) at 300 K. For example, for copper,
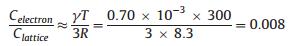
|
(1) |
where R is the gas constant and γ is the Sommerfeld's constant [0.70 mJ mol-1 K-2) for Cu]. When heat is deposited from exothermic surface reactions or photon flux, electrons heat up much faster (femtoseconds) than the lattice (picoseconds) because their heat capacity is much lower.
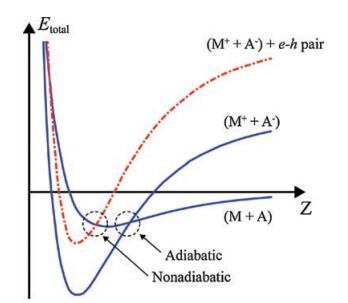
The phenomenon of electron exoemission from highly exothermic reactions of alkali metals with various gases is an indication that the gas-surface system cannot effectively dissipate energy by generating heat and is therefore described in the framework of a nonadiabatic mechanism, as illustrated in Fig. 1. Here, onedimensional potential energy curves (PECs) are shown for the cases of adiabatic and nonadiabatic interactions of a gas molecule with an atom on the metal surface. Such PECs are widely used in studies of the nonadiabatic phenomena that arise from chargetransfer reactions between reactants with very different electronegativities [28, 29]. They can also be used to describe the dissociative chemisorption of diatomic molecules on metal surfaces [21, 39]. A gas molecule (A) approaches an atom on a metal surface (M) so that their interaction is represented by the ground-state PECs before (M + A) and after (M+ + A-) charge transfer. As shown in Fig. 1, the reaction may proceed either adiabatically or nonadiabatically. In the latter case, the transition of the system to a new state occurs at the point of intersection of the PEC for (M + A) with one of the electronically excited states for (M+ + A-), which leads to the creation of an e-h pair. The subsequent relaxation of the system to the ground state can occur by emission of a photon or an exoelectron [19, 20, 28, 29].

|
Download:
|
| Fig. 1. One-dimensional PECs for the cases of adiabatic and nonadiabatic interactions of a gas molecule (A) with an atom on the metal surface (M). Adapted with permission [38]. Copyright 1991, AIP Publishing. | |
{kind=link}
Unlike adiabatic dissipation of chemical energy, which is characterized by the excitation of many phonons per elementary surface reaction, nonadiabatic reactions on metal catalysts most likely occur with the creation of a single e-h pair per exothermic reaction step [20, 28]. This is due to the continuity of the electronic levels in metals, which allows the transfer of essentially any portion of chemical energy to the electronic system with a single excitation. Relaxation of one e-h pair happens on the femtosecond to picosecond timescale, and its mean free path is on the order of tens of nanometers. Therefore, hot charge carriers created during transient processes (e.g., gas adsorption) decay rapidly when transferred through the metal-support interface; thus, the whole system goes to an equilibrium state [28, 40, 41]. However, during a stationary exothermic reaction when charge carrier excitation occurs because of a multitude of elementary reactions taking place on the catalyst per unit time, a steady-state hot electron (or hole) flux can be generated [2, 15, 20, 42]. Because hot electrons can transfer large amounts of energy, this flux can contribute significantly to the energy exchange process in the gas-surface system and thereby influence the reactivity of the catalyst [21, 28, 39, 43].
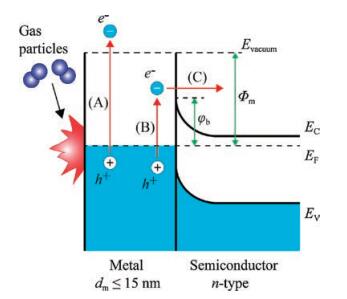
It is important to consider the question of how to detect hot charge carriers that are excited in metal catalysts by an exothermic chemical reaction. There are two detection strategies: The first is to obtain sufficient time resolution for observing these excitations. The second is to use a nanometer-scale energy barrier for irreversible transfer of hot electron fluxes. For the first approach, two-photon time-resolved photoemission (TPTRP) spectroscopy has been employed to directly study the dynamics of optically excited electrons at metal and semiconductor surfaces. This technique has been applied to the direct measurement of hot electron relaxation in noble and transition metals [44] and surfacestate dynamics on clean and adsorbate-covered metal surfaces [45], as well as charge carrier dynamics in semiconductors, where much work has been performed. As mentioned above, in the case of highly exothermic reactions taking place on the surface of metals with a low work function, nonadiabatic reactions can be detected by observing chemiluminescence or the emission of excited electrons into a vacuum, as shown in Fig. 2. However, for conventional catalytic reactions, this approach does not work because the work function of most catalytic metals (e.g., Pt, Pd, Rh) is much larger than the excess energy released by the surface reaction. Therefore, it was unclear for a long time if low-energy chemical reactions create hot charge carriers in metal catalysts [28]. A solution to this problem was found quite recently by Somorjai et al., who proposed the use of thin-film Schottky diodes, called catalytic nanodiodes, for detecting of hot electrons during catalytic reactions [15]. The proposed concept of hot electron detection is shown schematically in Fig. 2. An exothermic chemical reaction catalyzed on a metal surface creates a distribution of hot charge carriers (electrons and holes) located between levels corresponding to the Fermi energy in the metal and the maximal chemical energy liberated by the reaction (Eex). To detect hot carriers, a catalytic nanodiode can be used that is composed of an ultra-thin film of a catalytic metal deposited onto a semiconductor or insulator substrate. Here, excited electrons can be detected because of internal emission through the Schottky barrier formed at the metal-semiconductor interface. Since the thickness of the metal film is on the order of the mean free path of the hot electrons (i.e., typically ≤ 15 nm), these excited charge carriers can cross the film without significant attenuation and reach the metal-semiconductor interface while still energetic enough to overcome the Schottky barrier. The resulting chemicurrent, which is a flow of hot electrons, is proportional to the turnover frequency (TOF). Thus, the magnitude of the chemicurrent may be written as [19, 20, 46]:

|
Download:
|
| Fig. 2. Schematic layout and energy-band diagram of an experiment for detecting hot electrons created during a chemical reaction. Here, (A) is exoelectron emission (Eex >Φm), (B) is excitation, and (C) internal emission of a low-energy hot electron (φb < Eex < Φm), where Φm is the metal work function and φb is the Schottky barrier height. Adapted with permission [20]. Copyright 2016, Elsevier. | |
{kind=link}
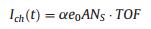
|
(2) |
where α is the chemicurrent yield, e0 is the electron charge, A is the surface area, and NS is the number of catalytic sites. Eq. (2) shows that the simultaneous measurement of chemical kinetics and chemicurrent makes it possible to estimate the probability of nonadiabatic reactions and determine their role in catalysis.
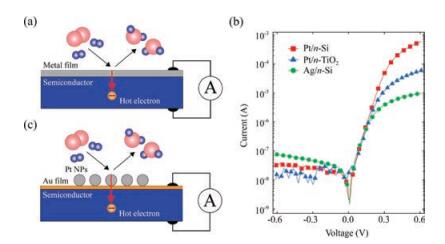
3. DeviceIn earlier experiments on the detection of hot electrons during catalytic chemical reactions, planar Schottky nanodiodes consisting of an ultra-thin Pt film deposited on an n-TiO2 or n-GaN substrate were used [15, 16, 47, 48]. Prior to these experiments, Schottky diodes with a similar architecture were also used by McFarland et al. in high-vacuum experiments aimed at detecting e-h pairs excited during the chemisorption of atomic hydrogen on the surface of Ag thin films supported on n-Si and p-Si substrates [40, 49]. A generalized scheme of the planar catalytic nanodiode is shown in Fig. 3a. A thin metal film acts as both the catalyst and electrode, thus allowing the flux of chemically excited hot electrons to be measured. To ensure a reliable supply of hot carriers from the surface of the film to the Schottky contact, the film thickness should not exceed the ballistic mean free path of electrons in the metal [50].

|
Download:
|
| Fig. 3. (a) Detection of chemically excited hot electrons based on a planar metalsemiconductor Schottky diode (catalytic nanodiode). (b) Typical current-voltage characteristics of commonly used catalytic nanodiodes. (c) Scheme of hot electron detection during a catalytic reaction on Pt nanoparticles supported on a catalytic nanodiode. | |
{kind=link}
Since the detection of hot electrons occurs during their transfer through the Schottky contact, a necessary condition for the operation of a catalytic nanodiode is a properly sized Schottky barrier [20, 28]. The height of the barrier must be high enough to limit the transfer of thermal carriers through the metalsemiconductor interface, thus allowing the detection of only hot charge carriers excited in the nonadiabatic reactions. However, the Schottky barrier must not exceed the exothermicity of the surface reaction. Thus, considerations for the transparency of the barrier for charge carriers with a certain excess energy should be taken into account when choosing a semiconductor support for the nanodiodes [20, 51]. For an ideal Schottky diode, the height of the barrier can be found as φb =Φm -χs, where Φm is the metal work function and χs is the electron affinity of the semiconductor [52]. However, for real nanodiodes, significant deviations from this equation can be observed. Thus, as a rule, the height of the barrier is determined by fitting an experimentally measured currentvoltage curve to the diode equation:
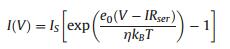
|
(3) |
where 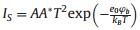
The use of planar Schottky nanodiodes have made it possible to carry out proof-of-concept experiments demonstrating the feasibility of studying nonadiabatic processes in catalytic reactions using solid-state devices. Nevertheless, the structure of such thin film nanodiodes differs significantly from the supported metalsemiconductor catalysts used in industrial processes. To fill this gap, several research groups have recently attempted to experiment with more complex nanodiodes whose structures simulate real-world catalysts. For instance, Park et al. demonstrated the possibility of detecting nonadiabatic electronic excitation in colloid nanoparticles (NPs) of platinum using the catalytic H2 and CO oxidation reactions [17, 46]. The idea of this experiment is clear from Fig. 3c: Pt NPs of a defined size are deposited as twodimensional arrays on an Au thin film that is supported on the TiO2 surface of the nanodiode. The Au film creates an electrical connection between the Pt NPs and the external circuit, thus allowing the chemicurrent to be measured. In addition, it prevents any interaction of the TiO2 layer with the reactive gas environment and helps to avoid any side effects associated with the adsorption of gas particles on the TiO2 (e.g., internal and external diffusion, spillover).
Another idea for making nanodiodes that simulate real catalysts was proposed by Schierbaum et al. who studied the effects of charge carrier creation during catalytic reactions on Pt/TiO2 nanodiodes based on porous titanium oxide layers fabricated using plasma electrolytic oxidation (PEO) of a Ti metal foil [53, 54]. Karpov et al. also conducted chemicurrent studies using similar nanodiodes consisting of a Pt mesh supported on mesoporous TiO2 and ZrO2 layers [55-57].
4. Applications 4.1. Gas-phase reactionsTo date, most studies of nonadiabatic reactions using Schottky diodes have used the catalytic hydrogen oxidation reaction [41, 46, 48, 55, 56, 58-60]. This is primarily because this reaction has a tremendous significance in both science and industry. As expected, nonadiabatic electronic excitation is also more frequent when light atoms and molecules interact with metallic surfaces, as is the case for surface reactions involving hydrogen [28, 43, 61].
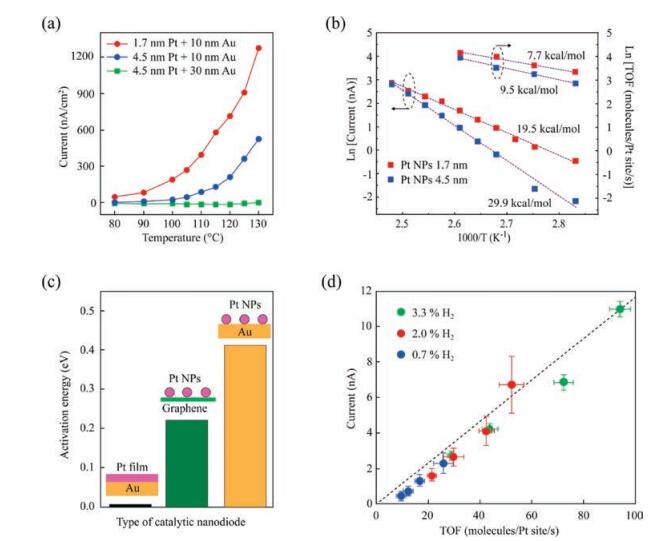
A typical chemicurrent signal that corresponds to the transfer of hot electrons excited by the catalytic reaction in a H2 (15 Torr) + O2 (745 Torr) gas mixture on 1.7 nm Pt NPs@Au/TiO2 nanodiodes is shown in Fig. 4a [46]. The chemicurrent demonstrates a noticeable temperature dependence that is caused by an increase in the rate of H2 oxidation at the elevated temperature of the Pt catalyst. The chemicurrent value is also dependent on the metal thickness. As can be seen, an increase in both Pt NPs size and Au film thickness leads to a sharp decrease in the chemicurrent, which is associated with a decrease in hot charge carrier flux because of electronelectron and electron-phonon scattering [46].

|
Download:
|
| Fig. 4. (a) Typical chemicurrent signals attributed to the transfer of hot electrons excited by catalytic H2 oxidation on Au/TiO2 nanodiodes decorated with two sizes of Pt nanoparticles [46]. (b) Arrhenius plots obtained from chemicurrent and TOF measurements during H2 oxidation on Au/TiO2 nanodiodes decorated with 1.7 nm and 4.5 nm Pt nanoparticles [46]. (c) Potential barrier heights at different Pt/support interfaces obtained from the Arrhenius plots for chemicurrent [60]. (d) Chemicurrent as a function of TOF for H2 oxidation, measured at different hydrogen concentrations [60]. Adapted with permission [46, 60]. Copyright 2015, Wiley-VCH. Copyright 2016, ACS Publications. | |
{kind=link}
The activation energy can be determined using the TOF and chemicurrent values measured as a function of temperature [2, 46, 48]. Early experiments on hot electron detection using planar Schottky nanodiodes showed identical activation energies for both the TOF and the chemicurrent [2, 16, 48]. For example, in ref. [48], an activation energy of Ea = 7.6 ± 0.6 (kcal/mol) is obtained from TOF measurements for H2 on the surface of Pt/TiO2 nanodiodes. This value coincides with the activation energy of Ea = 7.4 ± 0.3 (kcal/mol) determined from chemicurrent measurements, which implies that the chemicurrent originates from the catalytic reaction. However, in later studies detecting chemicurrents using catalytic nanodiodes decorated with Pt NPs, it was shown that the activation energy obtained from chemicurrent measurements can be significantly larger than that determined from TOF measurements [17, 46]. Typical Arrhenius plots demonstrating this trend are shown in Fig. 4b. The difference between activation energies obtained from the chemicurrent and TOF measurements also depends on the size of Pt NPs and the type of capping agent used during deposition of the NPs [20, 46]. Therefore, this effect is attributed to the peculiarities of hot electron transport across the nanoparticle/support interface.
Graphene has several extraordinary properties that make it a promising candidate for use as a catalytic support [60, 62-64]. Graphene is a particularly good conductor of ballistic electrons, and is thus an interesting material for detecting nonadiabatic electronic excitation in catalytic reactions using Schottky nanodiodes [60, 65-67]. To study the chemicurrent effect on Pt NPs supported on a graphene layer, a novel catalytic nanodiode was developed by Lee et al. [60]. The structure of this nanodiode is similar to the one shown in Fig. 3c. However, a single graphene layer deposited on a TiO2 film is used instead of gold for the electrical connection with the Pt NPs. The use of a graphene layer allows for a significant reduction in the interfacial resistance between the Pt NPs and the support. As shown in Fig. 4c, the potential barrier at the Pt NPs/graphene interface (5.1 kcal/mol) is significantly smaller than that for the Pt NPs/Au interface of the Pt NPs@Au/TiO2 nanodiodes (9.5kcal/mol) reported in earlier studies. Therefore, as shown in Fig. 4d, the chemicurrent measured on graphene-based catalytic nanodiodes increases linearly with increasing reaction rate under H2 oxidation, which indicates a significant improvement in performance when compared with the Pt NPs@Au/TiO2 nanodiode. Based on these results, the use of highly conductive materials based on graphene layers can significantly accelerate the transfer of charge and energy through the metal nanoparticle/support boundary, which has a positive effect on the catalytic properties of the system [60].
We note that the strong correlation between hot electron flux and catalytic activity, as shown in Fig. 4d, indicates that hot electron flux can also drive chemical reactions. This concept, recently demonstrated in Pt/CdSe/Pt nanodumbbells and Pt/GaN and Au/CeO2 metal-semiconductor structures [23, 68, 69], suggests a new method for electronic control of catalytic reactions.
Porous materials are of great importance in heterogeneous catalysis. Due to the large surface area, the peculiarities of their structure, and their unique diffusion properties, the use of porous materials as a support for metal catalysts leads to significant economic benefits [3, 70, 71]. To study the role of electronic excitation on the catalytic properties of porous materials, Schierbaum et al. and later Karpov et al. studied the effects of chemicurrent generation in catalytic nanodiodes based on mesoporous Pt/TiO2 and Pt/ZrO2 structures [53-57]. Unlike planar nanodiodes, nanodiodes based on porous materials exhibit a very high chemicurrent, which can be one or two orders of magnitude larger than chemicurrents measured from thin-film nanodiodes under similar reaction conditions. However, the mechanism for generating such a high chemicurrent is most likely associated with the proton-generation process resulting from the spillover effect, and not from the creation of hot electrons [54, 57]. Consequently, the diffusion of protons on the surface of the porous materials and inside the pores leads to the appearance of a large chemicurrent.
4.2. Liquid-phase reactionsAs shown in Section 3, the detection of nonadiabatic reactions using catalytic nanodiodes occurs because of the internal emission of hot electrons through the Schottky barrier and therefore does not depend of the density of the reacting medium. This provides an opportunity to study catalytic reactions occurring at liquid/solid interfaces that are of great practical importance for heterogeneous catalysis [42, 72-74]. To date, most data from the experimental detection of chemically excited hot electrons during liquid-phase reactions have been reported for the catalytic decomposition reaction of hydrogen peroxide (2H2O2catalyst 2H2O + O2), which is commonly used in industry as an oxidizing agent for pulp and paper bleaching, wastewater treatment, textile production, chemical synthesis, and many other applications [75, 76]. In addition, the decomposition of H2O2 is highly exothermic (ΔH = -25.3 kJ/mol [74]) and can therefore serve as a model system for mechanistic studies of energy and charge transfer in catalytic reactions at liquid/solid interfaces.
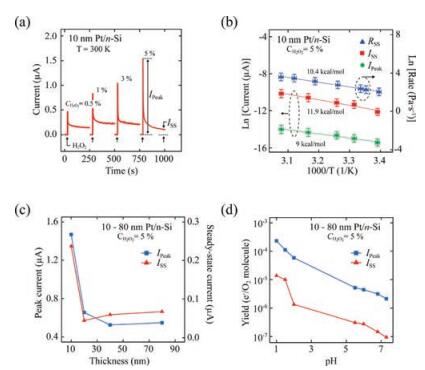
Typical chemicurrent signals measured on planar 10 nm Pt/n-Si nanodiodes in an aqueous solution containing H2O2 are shown in Fig. 5a. The chemicurrent is measured when the nanodiode is immersed into the H2O2 solution by the method described in ref. [42, 73]. The shape of the chemicurrent signal reflects the surface reaction rate. Initially, when the reaction rate is the greatest, the chemicurrent reaches a peak value. Then, as the steady-state reaction is established, the chemicurrent decreases and acquires a value that varies only little with time. The value of this steady-state chemicurrent is a clear function of the H2O2 concentration in the solution [73]. Thus, chemicurrent measurement provides an easyto-implement method for studying the catalytic process at the liquid/solid interface in real time.

|
Download:
|
| Fig. 5. (a) Time dependence of chemicurrent signals during the catalytic decomposition of H2O2 on 10-nm Pt/n-Si nanodiodes measured in solutions with different concentrations [73]. (b) Arrhenius plots obtained from measurements of chemicurrent and rate of oxygen evolution during the decomposition of H2O2 on 10- nm Pt/n-Si nanodiodes at different temperatures [73]. (c) Dependence of the chemicurrent on Pt film thickness [74]. (d) Chemicurrent measured on 10-nm Pt/nSi nanodiodes in H2O2 solutions with different pH values [74]. Adapted with permission [73, 74]. Copyright 2016, AIP Publishing. Copyright 2017, Elsevier. | |
{kind=link}
Activation energies can be determined by measuring the chemicurrent and the corresponding reaction rate at different temperatures [73]. Fig. 5b shows Arrhenius plots obtained for peak and steady-state chemicurrents (left scale) and the rate of O2 evolution (right scale) from the surface of 10 nm Pt/n-Si nanodiodes during the decomposition of a 5% aqueous H2O2 solution. The activation energy determined from the slope of the curves is Ea=10.4 ±0.3 (kcal/mol) for the reaction rate and Ea= 11.9 ±1.2 (kcal/mol) for the steady-state chemicurrent. These values are in a good agreement, which indicates that the chemicurrent is derived from the catalytic process. The activation energy obtained for the peak chemicurrent, Ea= 9.0 ± 0.7 (kcal/mol), is somewhat smaller than the value measured for the steady-state chemicurrent. The difference between the activation energy of the peak and steadystate chemicurrents can be related to the fact that these currents are attributed to different steps in the surface reaction [73].
The direction of the chemicurrent corresponds to the motion of hot electrons from the Pt film into the Si support. As shown in Fig. 5c, the magnitude of the chemicurrent decreases rapidly when the Pt film thickness increases because of the attenuation of hot electron flow in the Pt film by electron-electron and electronnuclei scattering [42, 74]. This dependence confirms that the chemicurrent measured during decomposition of H2O2 is from the hot electron effect.
The decomposition of H2O2 is an electrochemical reaction by nature. In solutions with high acidity, the reaction proceeds through the Pt-catalyzed dissociation of H2O2 molecules on the OH species (i.e., H2O2 +e-Pt OH + OH-) [77, 78]. The subsequent chain reactions lead to the formation of water and oxygen. Supposedly, the process of OH formation on the Pt surface is responsible for the creation of hot electrons [74]. In neutral and basic hydrogen peroxide solutions, however, H2O2 molecules can decompose directly to water and oxygen following the reaction 2H2O2catalyst 2H2O + O2 [77-80]. Therefore, it is of great interest to study the chemicurrent effect in H2O2 solutions with different pH values. In neutral and weakly acidic H2O2 solutions, the chemicurrent signal is low and almost independent of the pH, while in solutions with high acidity, the chemicurrent rapidly increases [74]. The rate of O2 evolution, however, shows the opposite trend when the pH value of the H2O2 solution varies. Thus, the chemicurrent yield, which is the number of hot electrons detected per O2 molecule formed on the nanodiode surface, increases exponentially with increasing acidity of the solution, as shown in Fig. 5d. This dependence is direct evidence of the more intensive charge transfer between the Pt surface and H2O2 molecules when the solution is highly acidic, leading to the selective generation of hot carriers with the appearance of only certain intermediate reaction products.
5. Concluding remarks and future direction of hot electron chemistryThe future direction of hot electron studies involves the development of an advanced scheme for hot electron detection using hybrid catalytic nanodiodes with a high surface area and in situ surface characterization of charge transport through metaloxide interfaces. In this approach, the Schottky barrier between metal nanoparticles or nanodisks on a semiconductor support has been probed with conductive probe atomic force microscopy [81, 82]. Experimental efforts to probe hot electrons enhanced by surface plasmons are currently in progress. A more fundamental understanding of hot electron phenomena would be required on both theoretical and experimental levels. Hot electrons on a metallic surface can be created by external energy deposition in the form of photons, ions, electrons, and chemical reactions. Therefore, we can consider hot electrons to be a major mediator for general energy conversion. The scheme of energy conversion from chemical (i.e., catalytic reactions) and photon-to-electrical energy (i.e., hot electron current) may give insight into other types of energy sources, including solar cells and electrochemical and photocatalytic devices.
In this review, we highlighted recent studies aimed at understanding the nature of charge transfer arising during catalytic reactions on supported metal catalysts. First, we discussed the basic mechanisms of chemical energy dissipation, including both adiabatic and nonadiabatic effects, that could be potentially responsible for creating hot charge carriers in metal catalysts during exothermic reactions. We also provided an overview of the main aspects related to the detection of electronic excitation during nonadiabatic energy dissipation in the form of hot electron flow using catalytic nanodiodes, which consist of nanostructured catalytically active metals deposited onto the surface of a semiconductor. We then showed that the detection of hot electrons in the form of chemicurrent allows for operando studies of nonadiabatic effects during gas/solid and liquid/solid reactions. Thus, it can be used as a powerful tool for the advanced study of heterogeneous reactions with energy release.
AcknowledgmentThis work was supported by the Institute for Basic Science (IBS, Republic of Korea) (No. IBS-R004-A2-2017-a00).
| [1] |
G.A. Somorjai, J.Y. Park, Angew. Chem. Int. Ed. 47 (2008) 9212-9228. DOI:10.1002/anie.200803181 |
| [2] |
J.Y. Park, Current Trends of Surface Science and Catalysis[M]. New York: Springer-Verlag, 2014.
|
| [3] |
J.M. Thomas, ChemSusChem 7 (2014) 1801-1832. DOI:10.1002/cssc.v7.7 |
| [4] |
C.M. Friend, B. Xu, Acc. Chem. Res. 50 (2017) 517-521. DOI:10.1021/acs.accounts.6b00510 |
| [5] |
G.A. Somorjai, H. Frei, J.Y. Park, J. Am. Chem. Soc. 131 (2009) 16589-16605. DOI:10.1021/ja9061954 |
| [6] |
J.A. Schwarz, C. Contescu, A. Contescu, Chem. Rev. 95 (1995) 477-510. DOI:10.1021/cr00035a002 |
| [7] |
P. Munnik, P.E. de Jongh, K.P. de Jong, Chem. Rev. 115 (2015) 6687-6718. |
| [8] |
J. Liu, ACS Catal. 7 (2017) 34-59. DOI:10.1021/acscatal.6b01534 |
| [9] |
G.T.K.K. Gunasooriya, E.G. Seebauer, M. Saeys, ACS Catal. 7 (2017) 1966-1970. DOI:10.1021/acscatal.6b02906 |
| [10] |
Y. Lykhach, S.M. Kozlov, T. Skala, Nat. Mater. 15 (2016) 284-288. DOI:10.1038/nmat4500 |
| [11] |
M.G. Willinger, W. Zhang, O. Bondarchuk, et al., Angew. Chem. Int. Ed. 53 (2014) 5998-6001. DOI:10.1002/anie.201400290 |
| [12] |
C.R. Henry, Surf. Sci. Rep. 31 (1998) 231-325. DOI:10.1016/S0167-5729(98)00002-8 |
| [13] |
G.M. Schwab, K. Koller, J. Am. Chem. Soc. 90 (1968) 3078-3080. DOI:10.1021/ja01014a016 |
| [14] |
G.M. Schwab, Surf. Sci. 13 (1969) 198-200. DOI:10.1016/0039-6028(69)90249-0 |
| [15] |
G.A. Somorjai, Catal. Lett. 101 (2004) 1-3. |
| [16] |
J.Y. Park, J.R. Renzas, B.B. Hsu, G.A. Somorjai, J. Phys. Chem. C 111 (2007) 15331-15336. DOI:10.1021/jp074562h |
| [17] |
J.Y. Park, H. Lee, J.R. Renzas, Y. Zhang, G.A. Somorjai, Nano Lett. 8 (2008) 2388-2392. DOI:10.1021/nl8012456 |
| [18] |
J.Y. Park, L.R. Baker, G.A. Somorjai, Chem. Rev. 115 (2015) 2781-2817. DOI:10.1021/cr400311p |
| [19] |
J.Y. Park, S.M. Kim, H. Lee, I.I. Nedrygailov, Acc. Chem. Res. 48 (2015) 2475-2483. DOI:10.1021/acs.accounts.5b00170 |
| [20] |
I.I. Nedrygailov, J.Y. Park, Chem. Phys. Lett. 645 (2016) 5-14. DOI:10.1016/j.cplett.2015.12.024 |
| [21] |
A.M. Wodtke, Chem. Soc. Rev. 45 (2016) 3641-3657. DOI:10.1039/C6CS00078A |
| [22] |
S. Mukherjee, F. Libisch, N. Large, et al., Nano Lett. 13 (2012) 240-247. |
| [23] |
S.M. Kim, S.J. Lee, S.H. Kim, et al., Nano Lett. 13 (2013) 1352-1358. DOI:10.1021/nl400367m |
| [24] |
C. Boerigter, U. Aslam, S. Linic, ACS Nano 10 (2016) 6108-6115. DOI:10.1021/acsnano.6b01846 |
| [25] |
S. Linic, U. Aslam, C. Boerigter, M. Morabito, Nat. Mater. 14 (2015) 567-576. DOI:10.1038/nmat4281 |
| [26] |
Z. Zhang, J.T. Yates Jr., Chem. Rev 112 (2012) 5520-5551. DOI:10.1021/cr3000626 |
| [27] |
S.W. Lee, C. Lee, K.C. Goddeti, S.M. Kim, J.Y. Park, RSC Adv. 7 (2017) 56073-56080. DOI:10.1039/C7RA10450B |
| [28] |
H. Nienhaus, Surf. Sci. Rep. 45 (2002) 1-78. DOI:10.1016/S0167-5729(01)00019-X |
| [29] |
T. Greber, Surf. Sci. Rep. 28 (1997) 1-64. DOI:10.1016/S0167-5729(97)00005-8 |
| [30] |
B. Kasemo, L. Walldén, Surf. Sci. 53 (1975) 393-407. DOI:10.1016/0039-6028(75)90139-9 |
| [31] |
B. Kasemo, E. Törnqvist, J.K. Nørskov, B.I. Lundqvist, Surf. Sci. 89 (1979) 554-565. DOI:10.1016/0039-6028(79)90637-X |
| [32] |
B. Kasemo, Phys. Rev. Lett. 32 (1974) 1114-1117. DOI:10.1103/PhysRevLett.32.1114 |
| [33] |
B. Kasemo, Surf. Sci. 363 (1996) 22-28. DOI:10.1016/0039-6028(96)00088-X |
| [34] |
T. Greber, R. Grobecker, A. Morgante, A. Böttcher, G. Ertl, Phys. Rev. Lett. 70 (1990) 1331-1334. |
| [35] |
T. Greber, K. Freihube, R. Grobecker, et al., Phys. Rev. B 50 (1994) 8755-8762. DOI:10.1103/PhysRevB.50.8755 |
| [36] |
D.N. Denzler, C. Frischkorn, C. Hess, M. Wolf, G. Ertl, Phys. Rev. Lett. 91 (2003) 226102. DOI:10.1103/PhysRevLett.91.226102 |
| [37] |
A. Böttcher, R. Imbeck, A. Morgante, G. Ertl, Phys. Rev. Lett. 65 (1990) 2035-2037. DOI:10.1103/PhysRevLett.65.2035 |
| [38] |
A. Böttcher, R. Grobecker, R. Imbeck, A. Morgante, G. Ertl, J. Chem. Phys. 95 (1991) 3756-3766. DOI:10.1063/1.460826 |
| [39] |
A.M. Wodtke, D. Matsiev, D.J. Auerbach, Prog. Surf. Sci. 83 (2008) 167-214. DOI:10.1016/j.progsurf.2008.02.001 |
| [40] |
B. Gergen, H. Nienhaus, W.H. Weinberg, E.W. McFarland, Science 294 (2001) 2521-2523. DOI:10.1126/science.1066134 |
| [41] |
S.K. Dasari, M.A. Hashemian, J. Mohan, E.G. Karpov, Chem. Phys. Lett. 553 (2012) 47-50. DOI:10.1016/j.cplett.2012.09.066 |
| [42] |
I.I. Nedrygailov, C. Lee, S.Y. Moon, H. Lee, J.Y. Park, Angew. Chem. Int. Ed. 128 (2016) 11017-11020. DOI:10.1002/ange.201603225 |
| [43] |
E. Hasselbrink, Curr. Opin. Solid State Mater. Sci. 10 (2006) 192-204. DOI:10.1016/j.cossms.2007.04.003 |
| [44] |
S. Ogawa, H. Petek, Surf. Sci. 363 (1996) 313-320. DOI:10.1016/0039-6028(96)00153-7 |
| [45] |
T. Hertel, E. Knoesel, M. Wolf, G. Ertl, Phys. Rev. Lett. 76 (1996) 535-538. DOI:10.1103/PhysRevLett.76.535 |
| [46] |
H. Lee, I.I. Nedrygailov, C. Lee, G.A. Somorjai, J.Y. Park, Angew. Chem. Int. Ed. 54 (2015) 2340-2344. DOI:10.1002/anie.201410951 |
| [47] |
X. Ji, A. Zuppero, J.M. Gidwani, G.A. Somorjai, Nano Lett. 5 (2005) 753-756. DOI:10.1021/nl050241a |
| [48] |
A. Hervier, J.R. Renzas, J.Y. Park, G.A. Somorjai, Nano Lett. 9 (2009) 3930-3933. DOI:10.1021/nl9023275 |
| [49] |
H. Nienhaus, H.S. Bergh, B. Gergen, et al., Surf. Sci. 445 (2000) 335-342. DOI:10.1016/S0039-6028(99)01078-X |
| [50] |
K.W. Frese, C. Chen, J. Electrochem. Soc. 139 (1992) 3234-3243. DOI:10.1149/1.2069059 |
| [51] |
H. Nienhaus, B. Gergen, W.H. Weinberg, E.W. McFarland, Surf. Sci. 514 (2002) 172-181. DOI:10.1016/S0039-6028(02)01625-4 |
| [52] |
S.M. Sze, K.K. Ng, Physics of Semiconductor Devices[M]. New York: John Wiley & Sons, 2006.
|
| [53] |
K. Schierbaum, M. Achhab, Phys Status Solidi A 208 (2011) 2796-2802. DOI:10.1002/pssa.201127400 |
| [54] |
Ö. Cakabay, M. El Achhab, K. Schierbaum, Appl. Phys. A 118 (2014) 1127-1132. |
| [55] |
E.G. Karpov, M.A. Hashemian, S.K. Dasari, J. Phys. Chem. C 117 (2013) 15632-15638. DOI:10.1021/jp402698f |
| [56] |
M.A. Hashemian, E. Palacios, I.I. Nedrygailov, D. Diesing, E.G. Karpov, ACS Appl. Mater. Interfaces 5 (2013) 12375-12379. DOI:10.1021/am403182v |
| [57] |
N.J. Ray, M.A. Hashemian, E.G. Karpov, ACS Appl. Mater. Interfaces 7 (2015) 27749-27754. DOI:10.1021/acsami.5b08814 |
| [58] |
E.G. Karpov, I.I. Nedrygailov, Appl. Phys. Lett. 94 (2009) 214101. DOI:10.1063/1.3147853 |
| [59] |
E.G. Karpov, I. Nedrygailov, Phys. Rev. B 81 (2010) 205443. DOI:10.1103/PhysRevB.81.205443 |
| [60] |
H. Lee, I.I. Nedrygailov, Y.K. Lee, et al., Nano Lett. 16 (2016) 1650-1656. DOI:10.1021/acs.nanolett.5b04506 |
| [61] |
E. Hasselbrink, Surf. Sci. 603 (2009) 1564-1570. DOI:10.1016/j.susc.2008.12.037 |
| [62] |
S. Guo, D. Wen, Y. Zhai, S. Dong, E. Wang, ACS Nano 4 (2010) 3959-3968. DOI:10.1021/nn100852h |
| [63] |
Y.P. Xiao, S. Wan, X. Zhang, et al., Chem. Commun. 48 (2012) 10331-10333. DOI:10.1039/c2cc35562k |
| [64] |
Y. Li, L. Tang, J. Li, Electrochem. Commun. 11 (2009) 846-849. DOI:10.1016/j.elecom.2009.02.009 |
| [65] |
W.K. Tse, E.H. Hwang, S.D. Sarma, Appl. Phys. Lett. 93 (2008) 023128. DOI:10.1063/1.2956669 |
| [66] |
X. Du, I. Skachko, A. Barker, E.Y. Andrei, Nat. Nanotechnol. 3 (2008) 491-495. DOI:10.1038/nnano.2008.199 |
| [67] |
A.S. Mayorov, R.V. Gorbachev, S.V. Morozov, et al., Nano Lett. 11 (2011) 2396-2399. DOI:10.1021/nl200758b |
| [68] |
S.M. Kim, D. Park, Y. Yuk, S.H. Kim, J.Y. Park, Faraday Discuss. 162 (2013) 355-364. DOI:10.1039/c2fd20133j |
| [69] |
S.M. Kim, H. Lee, K.C. Goddeti, S.H. Kim, J.Y. Park, J. Phys. Chem. C 119 (2015) 16020-16025. DOI:10.1021/acs.jpcc.5b03287 |
| [70] |
C. Perego, R. Millini, Chem. Soc. Rev. 42 (2013) 3956-3976. DOI:10.1039/C2CS35244C |
| [71] |
N. Pal, A. Bhaumik, RSC Adv. 5 (2015) 24363-24391. DOI:10.1039/C4RA13077D |
| [72] |
F. Zaera, Chem. Rev. 112 (2012) 2920-2986. DOI:10.1021/cr2002068 |
| [73] |
I.I. Nedrygailov, C. Lee, S.Y. Moon, H. Lee, J.Y. Park, Rev. Sci. Instrum. 87 (2016) 114101. DOI:10.1063/1.4967529 |
| [74] |
S.H. Lee, I.I. Nedrygailov, S. Oh, J.Y. Park, Catal. Today 303 (2018) 282-288. DOI:10.1016/j.cattod.2017.09.038 |
| [75] |
J.M. Campos-Martin, G. Blanco-Brieva, J.L.G. Fierro, Angew. Chem. Int. Ed. 45 (2006) 6962-6984. DOI:10.1002/(ISSN)1521-3773 |
| [76] |
N.M. Wilson, D.W. Flaherty, J. Am. Chem. Soc. 138 (2015) 574-586. |
| [77] |
J. Weiss, Trans. Faraday Soc. 31 (1935) 1547-1557. DOI:10.1039/tf9353101547 |
| [78] |
D.W. McKee, J. Catal. 14 (1969) 355-364. DOI:10.1016/0021-9517(69)90326-1 |
| [79] |
Y. Liu, H. Wu, M. Li, J.J. Yin, Z. Nie, Nanoscale 6 (2014) 11904-11910. DOI:10.1039/C4NR03848G |
| [80] |
Y. Su, Y. Ge, L. Liu, et al., ACS Appl. Mater. Interfaces 8 (2016) 4250-4257. DOI:10.1021/acsami.6b00012 |
| [81] |
H. Lee, Y.K. Lee, T.N. Van, J.Y. Park, Appl. Phys. Lett. 103 (2013) 173103. DOI:10.1063/1.4826140 |
| [82] |
S. Kwon, S.J. Lee, S.M. Kim, et al., Nanoscale 7 (2015) 12297-12301. DOI:10.1039/C5NR02285A |