 2018, Vol. 29
2018, Vol. 29 


 ,
Xingchuang Xiongb,
Tao Wangc,
Jinan Lia,
Fangjun Wanga,
Xiang Fangb,
Xueming Yangc
,
Xingchuang Xiongb,
Tao Wangc,
Jinan Lia,
Fangjun Wanga,
Xiang Fangb,
Xueming Yangc
b National Institute of Metrology, Beijing 100013, China;
c State Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
Phosphorylation is a crucial protein post-translational modification (PTM) and plays key regulatory roles in many vital biological processes, such as signal transduction, mitosis and cell differentiation. Usually, these biological processes are spatiotemporally regulated by sequential and multiple phosphorylation statuses with different phosphorylation sites [1-4]. The involvement of multi-phosphorylation sites could offer much more versatile and precise regulation of the protein functions and has gained more and more attentions in the recent years [5-8]. Unambiguous assignment of multiple phosphorylation sites is essential to elucidate the mechanisms of multi-phosphorylation regulating biological processes. Although conventional mutation-based biological method can definitely confirm the possible phosphorylation sites, it is time consuming, low throughput and expensive [9, 10]. Mass spectrometry (MS)-based phosphoproteomic technologies could characterize thousands of phosphopeptides to date. However, unambiguous assignment of the phosphorylation sites on protein sequences is still a big challenge, especially for protein sequences with multiple possible phosphorylation sites closed to each other [11-13]. The accuracy of phosphorylation site assignment is mainly dependent on the phosphorylation site-determining fragment ions in MS/MS analysis. Traditional collision induced dissociation (CID) and higher energy collision dissociation (HCD) strategies usually result a neutral loss of 98 Da in MS/MS analyses due to the loss of labile phosphate group in the slow-heating dissociation process [14, 15]. Recently developed electron transfer dissociation (ETD) and electron capture dissociation (ECD) are radical based reaction dissociation strategies and mainly generate c-, z-type fragment ions with less neutral loss. However, these reaction dissociation strategies are charge dependent, which shown poor efficiency of dissociation for the peptides with low charge state [16].
Ultraviolet photodissociation (UVPD) is a promising technology in elucidating the sequences and structures of large biomolecules [17, 18], such as proteins [19, 20], nucleic acids [21] and polysaccharides [22]. Through a fast deposit of high energy on the biomolecules by photons, UVPD could generate rich types of fragment ions, which could significantly improve the sequence coverage and structure elucidation accuracy of biomolecules [17, 18]. For example, b-, y-types of fragment ions are usually the dominant ones by traditional slow-heating CID and HCD in peptides and proteins sequencing, but UVPD can generate a-, x-, b-, y-, c-, z-types of fragment ions. The high-energy fast exciting UVPD strategy utilizing 193-nm ultraviolent laser can induce backbone dissociation of phosphopeptides to generate much more types of fragment ions with less influence on the side chain phosphate groups [11, 23]. Recently, it is demonstrated that the UVPD strategy could provide more information to generally improve the phosphorylation site localization accuracy in largescale phosphoproteomic analyses [11, 24]. However, the ability of UVPD to distinguish the phosphorylation site isomers of the multiphosphopeptides has been not systematically investigated until now.
In the present study, a 193-nm ultraviolet laser-MS system was established (Fig. S1 in Supporting information) and its potentiality in sophisticated multi-phosphorylation site elucidation was evaluated by using three groups of elaborate designed peptide isomers with various multi-phosphorylation statuses. These multiphosphorylated isomers were originated from the functional sequence regions of cell cycle regulating protein Sic1 [10, 25], transcriptional activator Gli3 [26] and microtubule-associated protein Tau [27, 28], which were all reported with multiphosphorylation regulated biological functions. We observed that even the subtle position changes of multiple possible phosphorylation sites could be accurately localized with high confidence by utilizing the UVPD strategy due to its ability in generating more types of fragment ions and eliminating the phosphate neutral loss. Comparing with conventional CID, the numbers of matched fragment ions and phosphorylation site-determining ions were averagely improved 123% and 104%, respectively. Our results demonstrated the usefulness of UVPD in accurately assignment the multi-phosphorylation sites within important phosphoproteins, which exhibited great potential in investigating the mechanisms of multi-phosphorylation co-regulating.
To validate the 193-nm ultraviolet laser-MS system established in this work, a standard peptide with a lysine residue on the ctermini (NIEDVIAQGIGK) was directly injected into the MS and dissociated by UVPD. Two charge states (+1, +2) of this peptide were clearly observed in the full MS spectra (Fig. S2 in Supporting information) and both were subjected to UVPD and conventional CID dissociation, respectively. Although the peptide sequence could be well elucidated by both UVPD and CID, the UVPD strategy produced 150% more fragment ions. All theoretical backbone fragmentation ion types could be observed consecutively in the UVPD spectra, including a, b, c, x, y and z ions plus a few of d, v, w ions (Fig. S2). The rich information of backbone fragment ions feasibly enhanced the confidence of peptide sequencing. Above results were consistent with the previous works [29, 30] and indicated that the ultraviolet laser-MS system constructed in this work was well worked in peptide fragmenting.
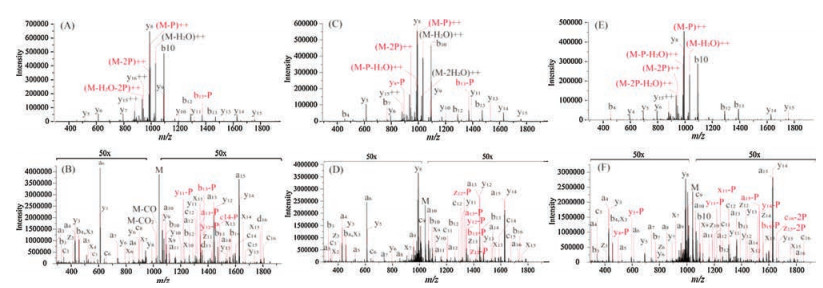
Unambiguously confirming the multiple possible phosphorylation sites closed to each other in the protein sequence is still a challenging task in MS analysis due to the neutral loss of phosphate group (98Da) and the lack of phosphorylation site-determining fragment ions [12]. As UVPD could induce backbone fragmentation to generate rich information of different types of fragment ions, it might be useful in improving the accuracy of multi-phosphorylation elucidating. Sic1, a key regulating protein in the cell cycle development, was reported to be destructed at the onset of S phase due to multi-phosphorylation in the sequence region of TPQKPSQNLVPVTPSTTK [25]. Thus, three multi-phosphopeptide isomers mimicking the multi-phosphorylation statuses of Sic1 were synthesized (pT13pT16, pT13pT17 and pS15pT16, Table S1 in Supporting information). The phosphorylation sites were designed to be closed to each other to challenge the capability of CID and UVPD in the accurate multi-phosphorylation elucidating. The CID spectra of the three multi-phosphorylated isomers were all dominated by the ions with neutral loss of phosphate group (-98 Da) and H2O (-18 Da). The intensity of typical CID fragment ions such as b-and y-type ions were relative low except for b10 and y8 (Figs. 1A, C and E), which is attributed to that the proline containing peptides were tend to yield abundant y-type fragment by cleaving the amide bond of proline residue [31]. Interestingly, the neutral loss of phosphate group was significantly suppressed in the UVPD spectra with relative abundance below 1%. More importantly, the backbone cleavages at the corresponding phosphorylation sites were feasibly achieved by UVPD to generate much more phosphorylation site-determining fragment ions (Figs. 1B, D and F). For example, the a-type ions in the UVPD spectra were consecutive and covered almost the whole peptides sequences. Owing to the higher sequences coverage and more phosphorylation site-determining ions provided by UVPD, all the phosphorylation sites could be precisely assigned onto the peptide sequences. However, only the phosphorylation site pT13 could be confidently assigned by all the CID spectra. Compared with the CID spectra, the numbers of matched fragment ions and phosphorylationsite-determiningionsobtainedintheUVPDspectra were averagely improved 1.7 and 1.6 times, respectively (Table S2 in Supporting information).

|
Download:
|
| Fig. 1. The MS/MS spectra of the Sic1 multi-phosphorylated isomers: TPQKPSQNLVPVpTPSpTTK (A, B), TPQKPSQNLVPVpTPpSTTK (C, D) and TPQKPSQNLVPVTPpSpTTK (E, F) by utilizing CID and UVPD strategies, respectively. | |
{kind=link}
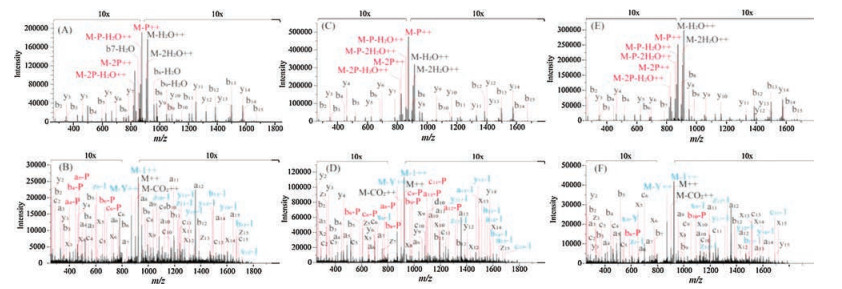
Then, the multi-phosphorylation region (RDSSASTISSAYLSSR) of Gli3, an important protein in regulating the embryonic development [26], was synthesized with various multiphosphorylation statuses (pS4pS6, pS6pS8 and pS6pS10, Table S1). Similar to the above results, the fragment ions originated from the neutral loss of phosphate group were greatly suppressed in the UVPD spectra and the backbone cleaving processes were dominated in the UVPD fragmentation. Briefly, the average numbers of matched fragment ions and phosphorylation sitedetermining ions were 55, 26 and 11, 6, from the UVPD and CID spectra of these multi-phosphorylated isomers, respectively (Fig. 2, Table S3 in Supporting information). However, additional 1 Da and 107 Da mass shifts from the precursor ions were observed in the UVPD dissociation of Gli3 isomeric group. It has been reported that tyrosine (Y) containing peptide would suffer a neutral loss of the tyrosine side chain and cause a 107 Da mass loss compared to the precursor ions [11, 32]. Thus, we speculated the existence of tyrosine residue in the sequences of Gli3 multiphosphorylated isomers might induce the mass defects of 1 Da and 107 Da, which could be attributed to the side chain loss of hydrogen and C7H7O· radicals (Fig. S3 in Supporting information), respectively. What's more, the loss of hydrogen and C7H7O· radicals were also observed in the consecutive tyrosine containing fragment ions (Figs. 2B, D and F). We also observed that the intensity of most of the x, y, z ions in Gli3 group spectra was relative low, compared to the Sci1 group isomers and the standard non-phosphorylated peptides. This might be attributed to the multiple side-chain fragmentation pathways suffered by the tyrosine containing fragment ions, which could disperse their intensities.

|
Download:
|
| Fig. 2. The MS/MS spectra of the Gli3 multi-phosphorylated isomers: RDSpSApSTISSAYLSSR (A, B), RDSSApSTIpSSAYLSS (C, D) and RDSSApSTISpSAYLSSR (E, F) by utilizing CID and UVPD strategies, respectively. | |
{kind=link}
The multi-phosphorylation region (SGYSSPGSPGTPGSR) of Tau, a crucial hyper-phosphorylated protein in the emergence and development of Alzheimer's disease [27, 28], was further synthesized and three different multi-phosphorylation statuses were designed (pY3pS5, pS5pS8 and pS4pS5, Table S1). Finally, the average numbers of matched fragment ions and phosphorylation sitedetermining ions were 36, 19 and 9, 6, in the UVPD and CID spectra of these three multi-phosphorylated isomers, respectively (Fig. 3, Table S4 in Supporting information). The precise localization of the phosphate groups was all achieved by UVPD strategy except for the pY3 site of pY3pS5 isomer, which was also a problem by CID. In the UVPD spectra of multi-phosphorylated isomers without pY (pS5pS8 and pS4pS5), the 1 and 107 Da mass loss from the precursor ions were observed and several fragment ions with a mass loss of 107 Da were also produced (Fig. 3F), which were consistent with the above results. However, none of these types of fragment ions were observed in the UVPD spectrum of pY3pS5 isomer. Therefore, phosphorylation might suppress of the dissociation of tyrosine side chain in UVPD.

|
Download:
|
| Fig. 3. The MS/MS spectra of the Tau multi-phosphorylated isomers: SGpYSpSPGSPGTPGSR (A, B), SGYSpSPGpSPGTPGSR (C, D) and SGYpSpSPGSPGTPGSR (E, F) by utilizing CID and UVPD strategies, respectively. | |
{kind=link}
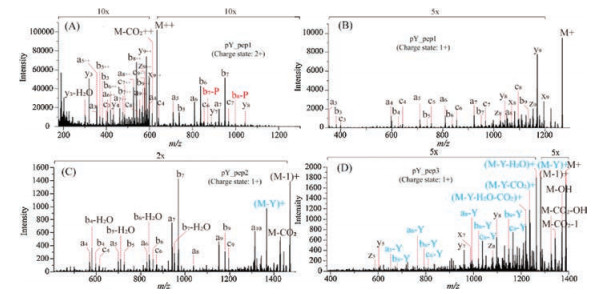
In order to conform the fragmentation behavior of the phosphorylated tyrosine residues in the UVPD, another threephosphorylated tyrosine (pY) containing peptides PQRpYLVIQGD, EPQpYEEIPIYL and YQHpYDLDLKD were synthesized and dissociated by UVPD (Fig. 4). The mass defects of 1 Da and 107 Da from the precursor ions were not observed in UVPD spectra of PQRpYLVIQGD (Figs. 4A and B). However, EPQpYEEIPIYL and YQHpYDLDLKD, which have another non-phosphorylated tyrosine within the peptides sequences, all generated 1 and 107 Da mass shifts in the UVPD spectra (Figs. 4C and D). The UVPD fragmentation pathway of the loss of tyrosine side chain was well established in YQHpYDLDLKD dissociation and the most abundant fragment ion was the M-107 Da (Fig. 4D). Thus, above results clearly demonstrated the phosphorylation of tyrosine could suppress the loss of its side chain in the UVPD dissociation.

|
Download:
|
| Fig. 4. The UVPD MS/MS spectra of the three pY containing peptides; (A) PQRpYLVIQGD (charge state 2+), (B) PQRpYLVIQGD (charge state 1+), (C) EPQpYEEIPIYL (charge state 1+) and (D) YQHpYDLDLKD (charge state 1+) | |
{kind=link}
Furthermore, the attenuation of UVPD fragmentation efficiency was observed for the pY containing isomer. The relative intensity of precursor ions in the total UVPD spectra was investigated for the three Tau group isomers, which could reflect the UVPD fragmentation efficiency to the precursor ions at identical condition (Fig. S4 in Supporting information). Obviously, the pY3pS5 isomer exhibited the lowest fragmentation efficiency among the three isomers. As the tyrosine side chain is a chromophore, the modified phosphate group may change the UV absorption property. It was reported that the deprotonated phosphate group with negative charge would induce blue shift of tyrosine adsorption wavelength and greatly suppressed its adsorption at 275 nm [33]. Thus, the fragmentation efficiency of these three multi-phosphorylated isomers in negative mode MS was also examined and the pY3pS5 isomer exhibited the highest fragmentation efficiency with 193-nm laser (Fig. S4), which was consist with the blue shift of tyrosine adsorption. The decrease of the fragmentation efficiency on positive mode MS might be attributed to the red shift of tyrosine adsorption cause by protonated phosphorylated group.
In this study, three groups of elaborate designed multiphosphorylated isomers, coming from the functional sequence regions of three important multi-phosphorylated proteins Sic1, Gli3 and Tau, were utilized to challenge the ability of multiphosphorylation statuses elucidation by MS with UVPD and CID strategies, respectively. For the total 9 multi-phosphopeptides with 18 phosphorylation sites difficult to be discriminated, the UVPD strategy precisely determined the positions of 17 sites (94%) with high confidence (the numbers of phosphorylation sitedetermining ions were averagely improved 104%). Obviously, the UVPD strategy could significantly improve the MS ability in elucidating the confused phosphorylation sites with multiple possible positions due to its ability in generating more phosphorylation site-determining ions and providing higher sequence coverage. However, due to the multiple fragmentation pathways of UVPD to generate more types of fragment ions, a dilution effect was observed in this work. The intensities of the UVPD fragment ions were relatively low compared to the corresponding CID spectra. What is more, the low resolution of LTQ MS caused the peak overlaps and the difficulty in peak assignment, which would be improved by using high resolution MS in the future work. Overall, the UVPD strategy is still a promising MS dissociation strategy and might play important roles in investigating the biological processes related with multi-phosphorylation coregulating.
AcknowledgmentsFinancial supports are gratefully acknowledged for the China State Key Research Grant (No. 2016YFF0200504), China State Key Basic Research Program Grant (No. 2013CB911203), the National Natural Science Foundation of China (No. 21675152), the Youth Innovation Promotion Association CAS (No. 2014164) and grant from DICP (No. ZZBS201603).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2017.10.020.
| [1] |
E.K. Lau, M. Trester-Zedlitz, J.C. Trinidad, et al., Sci. Signal. 4 (2011) ra52. |
| [2] |
M. Thomson, J. Gunawardena, Nature 460 (2009) 274-277. DOI:10.1038/nature08102 |
| [3] |
R.J. Deshaies, J.E. Ferrell Jr., Cell 107 (2001) 819-822. DOI:10.1016/S0092-8674(01)00620-1 |
| [4] |
P. Cohen, Trends Biochem. Sci. 25 (2000) 596-601. DOI:10.1016/S0968-0004(00)01712-6 |
| [5] |
C. Salazar, T. Hofer, FEBS J. 276 (2009) 3177-3198. DOI:10.1111/ejb.2009.276.issue-12 |
| [6] |
M. Vleugel, M. Omerzu, V. Groenewold, et al., Mol. Cell 57 (2015) 824-835. DOI:10.1016/j.molcel.2014.12.036 |
| [7] |
B.Y. Rubinstein, H.H. Mattingly, A.M. Berezhkovskii, S.Y. Shvartsman, Mol. Biol. Cell 27 (2016) 2331-2340. DOI:10.1091/mbc.e16-03-0137 |
| [8] |
J. Pan, S. Zhang, C.H. Borchers, J. Proteomics 134 (2016) 138-143. DOI:10.1016/j.jprot.2015.12.002 |
| [9] |
W.E. Borek, L.M. Groocock, I. Samejima, et al., Nat. Commun. 6 (2015) 7929. DOI:10.1038/ncomms8929 |
| [10] |
M. Koivomagi, M. Ord, A. Iofik, et al., Nat. Struct. Mol. Biol. 20 (2013) 1415-1424. DOI:10.1038/nsmb.2706 |
| [11] |
M.R. Robinson, J.M. Taliaferro, K.N. Dalby, J.S. Brodbelt, J. Proteome Res. 15 (2016) 2739-2748. DOI:10.1021/acs.jproteome.6b00289 |
| [12] |
S.A. Beausoleil, J. Villen, S.A. Gerber, J. Rush, S.P. Gygi, Nat. Biotechnol. 24 (2006) 1285-1292. DOI:10.1038/nbt1240 |
| [13] |
H. Wiese, K. Kuhlmann, S. Wiese, et al., J. Proteome Res. 13 (2014) 1128-1137. DOI:10.1021/pr400402s |
| [14] |
W.D. Lehmann, R. Kruger, M. Salek, et al., J. Proteome Res. 6 (2007) 2866-2873. DOI:10.1021/pr060573w |
| [15] |
A.M. Palumbo, S.A. Smith, C.L. Kalcic, et al., Mass Spectrom. Rev. 30 (2011) 600-625. DOI:10.1002/mas.v30.4 |
| [16] |
H. Molina, D.M. Horn, N. Tang, S. Mathivanan, A. Pandey, Proc. Natl. Acad. Sci. U. S. A. 104 (2007) 2199-2204. DOI:10.1073/pnas.0611217104 |
| [17] |
J.P. Reilly, Mass Spectrom. Rev. 28 (2009) 425-447. DOI:10.1002/mas.v28:3 |
| [18] |
A. Giuliani, A.R. Milosavljevic, F. Canon, L. Nahon, Mass Spectrom. Rev. 33 (2014) 424-441. DOI:10.1002/mas.v33.6 |
| [19] |
J.B. Shaw, W. Li, D.D. Holden, et al., J. Am. Chem. Soc. 135 (2013) 12646-12651. DOI:10.1021/ja4029654 |
| [20] |
J.P. O'Brien, W. Li, Y. Zhang, J.S. Brodbelt, J. Am. Chem. Soc. 136 (2014) 12920-12928. DOI:10.1021/ja505217w |
| [21] |
S.I. Smith, J.S. Brodbelt, Anal. Chem. 82 (2010) 7218-7226. DOI:10.1021/ac100989q |
| [22] |
B.J. Ko, J.S. Brodbelt, Anal. Chem. 83 (2011) 8192-8200. DOI:10.1021/ac201751u |
| [23] |
J.E. Mayfield, M.R. Robinson, V.C. Cotham, et al., ACS Chem. Biol. 12 (2017) 153-162. DOI:10.1021/acschembio.6b00729 |
| [24] |
K.L. Fort, A. Dyachenko, C.M. Potel, et al., Anal. Chem. 88 (2016) 2303-2310. DOI:10.1021/acs.analchem.5b04162 |
| [25] |
M. Koivomagi, E. Valk, R. Venta, et al., Nature 480 (2011) 128-131. DOI:10.1038/nature10560 |
| [26] |
Y. Chen, J. Jiang, Cell Res. 23 (2013) 186-200. DOI:10.1038/cr.2013.10 |
| [27] |
K. Blennow, H. Hampel, Lancet Neurol. 2 (2003) 605-613. DOI:10.1016/S1474-4422(03)00530-1 |
| [28] |
L. Buée, T. Bussière, V. Buée-Scherrer, A. Delacourte, P.R. Hof, Brain Res. Rev. 33 (2000) 95-130. DOI:10.1016/S0165-0173(00)00019-9 |
| [29] |
J.A. Madsen, T.S. Kaoud, K.N. Dalby, J.S. Brodbelt, Proteomics 11 (2011) 1329-1334. DOI:10.1002/pmic.201000565 |
| [30] |
J.H. Moon, S.H. Yoon, M.S. Kim, Rapid Commun. Mass Spectrom. 19 (2005) 3248-3252. DOI:10.1002/(ISSN)1097-0231 |
| [31] |
T. Vaisar, J. Urban, J. Mass Spectrom. 31 (1996) 1185-1187. DOI:10.1002/(ISSN)1096-9888 |
| [32] |
J. Lemoine, T. Tabarin, R. Antoine, M. Broyer, P. Dugourd, Rapid Commun. Mass Spectrom. 20 (2006) 507-511. DOI:10.1002/(ISSN)1097-0231 |
| [33] |
N. Okishio, R. Fukuda, M. Nagai, et al., J. Raman Spectrosc. 29 (1998) 31-39. DOI:10.1002/(ISSN)1097-4555 |