 2018, Vol. 29
2018, Vol. 29 



b School of Life Science and Engineering, Southwest Jiaotong University, Chengdu 611756, China
Protein-protein interactions (PPIs) play a central role in most biological processes and have been demonstrated to be important therapeutic targets in the past decades [1]. Synthetic peptides are suitable targeting candidates for PPIs as they are able to precisely mimic the topological features of target proteins and can be easily synthesized and modified [2]. Chemists have elaborated many efforts in order to stabilize these targeting epitopes into varies conformational elements, such as α-helix and β-hairpin [3]. Particularly, over 30% of protein secondary structure are helical in nature and over 50% of PPIs involve short a-helices according to a statistical analysis of protein data bank (PDB) [4]. As α-helix involved in a majority of PPIs, chemical methods, such as disulfide formation [3j], amide formation [3c], olefin/alkyne methathesis [3a], cysteine alkylation [3k] and perfluoroated arene incorportation [3l] have been utilized to confine peptides into helical structures.
Our group recently discovered a unique phenomenon [3d] that a precisely positioned chiral center in the tether of short peptides could dominantly induce helicity of the backbone peptides. This phenomenon could be extended to both a chiral sulfilimine center [5] and a chiral sulfoxide center [6] (Fig. 1), which provides a valuable modifiable site on the tether [7]. The absolute configuration of the in-tether chiral center was determined to be R for helicity inducement by peptide crystal structure.

|
Download:
|
| Fig. 1. Schematic illustration of reversibly switching of peptide conformation through an in-tether chiral sulfonium. | |
Methionine alkylation is a facile and chemoselective approach for introducing functional groups into peptides [8]. Deming et al. reported a chemoselective tagging strategy for the modification of peptides in a reversible manner [9]. Met residues could be chemoselectively alkylated in quantitative yields under acidic condition with satisfying residue tolerances [10]. Inspired by the versatility of methionine alkylation on especially its great functional group tolerance even for residues like Lys and Cys, we propose a simple and versatile strategy for reversibly controlling the conformation of short α-helical peptides. A thioether tethered peptide could be facially synthesized via photo-induced intramolecular thiol-ene reaction but too flexible to induce peptide's helicity [11]. We could then functionalize this thioether tether to generate a sulfonium based chiral auxiliary by a facile alkylation of the tether with alkyl halides under acidic media of trifluoroacetic acid (TFA) [12]. We envision the newly-generated sulfonium chiral center could modulate the backbone peptides' secondary structure, similarly as the all-hydrocarbon, sulfilimine and sulfoxide chiral centers. More importantly, the detachment and recovery of the parent thioether tethered stapled peptides could possibly enable the conformation switching between random coil and α-helix (Fig. 1).
Model peptide Ac-Y-(cyclo-1, 5)-mS5AAAC-NH2 was synthesized through intramolecular thiol-ene reaction (Fig. 2A) [11a].Then the cleaved peptide was let react with a panel of different alkylation reagents (Fig. 2B) in TFA at room temperature. We chose alkylating reagents in terms of their sizes and functional groups. Propinyl and allyl group were successfully attached to the peptide by alkyaltion reagents (Ⅱ) and (Ⅲ) respectively, which might enable a further bioorthogonal modifications [13]. Methyl group of (Ⅳ) and the aromatic groups benzyl of (Ⅴ), 4-fluorobenzyl of (Ⅵ) and 4-acetoxylbenzyl of (Ⅶ) were also tested as alkylation groups by there corresponding alkylation reagents [14]. The alkylation reaction occurred smoothly over 48 h in quantitive conversions except for aliphatic group of allyl and methyl. Two products of identical molecular weight but different HPLC retention time were identified, which represented two sulfonium epimers, similar as in our previous reports [3d]. The two epimers of peptides 2a/2b–7a/7b (Table 1) could be readily separated by reverse-phase HPLC (Fig. 2C, Figs. S3–S8 in Supporting information). The ratio of the two epimers was approximately 1:1 according to HPLC traces in Figs. S3–S8. The electron withdrawing vinyl and benzyl groups tended to accelerate the alkylation process. The retention time differences of the peptide epimers indicated a significant conformational difference between the two epimers.

|
Download:
|
| Fig. 2. (A) Alkylation of thioether tethered peptides in TFA at room temperature (1 mg/mL, alkylation reagent: 10.0 equiv.). (B) Alkylation reagents used in this study and alkylation conversions. (C) HPLC separation spectrum of alkylated peptide epimers 5a and 5b. | |
|
|
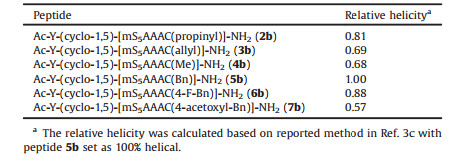
Table 1 Peptide sequences used in this study and relative helicity. |
{kind=link}
{kind=link}
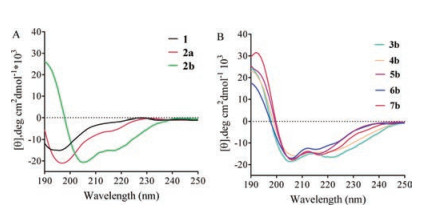
The peptide epimers' conformational preferences were tested and relative helicity of 2b–7b epimers summarized in Table 1. Details of molar elipticities at λ = 215, 206 and 190 nm were summarized in Table S2 in Supporting information. Consistent with our previous report, the b-epimers with longer retention time showed helical features in circular dichroism (CD) spectroscopy study while the a-epimers was mainly random coil. As shown in Fig. 3A, epimer 2b displayed featured helical conformation with double minima at 206 nm and 215 nm [15]. Epimer 2a and its parent thioether tethered peptide 1 showed typical CD spectra of random coils. Sulfonium peptides with different alkylation groups showed similar pattern as peptide 2a and 2b. The CD spectra were summarized in Figs. 3A and B. CD spectra of 3a–7a epimers were shown in Fig. S1 in Supporting information.

|
Download:
|
| Fig. 3. (A) CD spectra of thioether tethered peptide 1 and sulfonium functionalized peptide 2a/2b in double distilled water. (B) CD spectra of chiral sulfonium induced α-helical peptides 3b–7b in 20% TFE. | |
{kind=link}
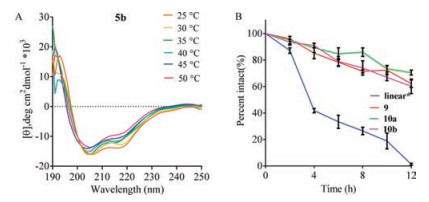
2D 1H NMR spectroscopy was conducted in H2O:D2O (9:1) at 298 K. We used peptide 10b (peptide 5b without concentration determining residue Tyr) as model peptide. The low amide coupling constants (3JNH-Hα < 6 Hz of three alanines) suggested a helical structure (Table S3) [3c, 16, 17]. However, due to the signal overlapping of protons, we were unable to unambiguously determine the key ROEs from NH(i)–Ha (i + 4). Then the thermal stability of 2b–7b epimers was tested using CD spectroscopy [18]. We observed gradual decreases of molar ellipticity as temperature increases from 25 ℃ to 50 ℃ (Fig. 4A, Fig. S2 in Supporting information). To further understand the cause of decrease in helicity of the sulfonium functionalized peptides, we incubated the peptide 8b (Ac-(cyclo-1, 5)-[mS5AYAC(4-F-Bn)]-NH2) under 50 ℃ overnight. We detected about 36% racemization of peptide 8b which indicated the decrease of helical content is attributed to the racemization of the chiral auxiliary (Fig. S12 in Supporting information). Meanwhile, the in vitro serum stability assay (Fig. 4B) showed that cyclic peptide 9 and the sulfonium peptides 10a and 10b remained more than 60 percent intact after 12 h, while the linear analogue was degraded within a few hours.

|
Download:
|
| Fig. 4. (A) CD spectra of 5b at elevated temperatures from 25 ℃ to 50 ℃. (B) In vitro serum digestion assay of peptides at 37 ℃ for 0–12 h. Percentage intact, mean ± s. d. and n = 3. * represent peptides without the residue Tyr. | |
{kind=link}
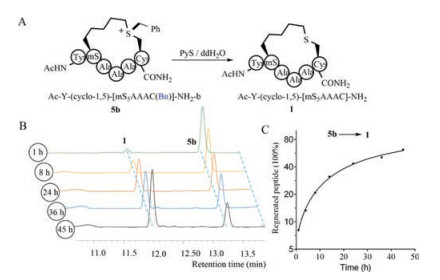
Furthermore, to explore the detachment of the sulfonium auxiliary, peptide 5b was chosen for model study (Fig. 5A). 2-Mercaptopyridine (PyS) was used as dealkylation reagent as reported previously [9]. PyS stock (100 mL, 1 mg/mL, 3 equiv.) was added to the aqueous solution of peptide 5b (0.6 mmol/L, 500 mL) at room temperature. HPLC traces were recorded at different time intervals as shown in Fig. 5B. The regenerated thioether-tethered peptide 1 was observed over time. Peptide 1 ca. 62% was regenerated during 48 h according to integration of the HPLC traces (Figs. 5B and C). Thus, the sulfonium chiral center could be successfully dealkylated to regenerate the parent

|
Download:
|
| Fig. 5. (A) Dealkylation of chiral sulfonium α-helical peptides in the presence of PyS in double distilled water. (B) HPLC traces of dealkylation process of 5b at different time intervals. (C) Plots of percent regenerated peptides versus time. | |
{kind=link}
thioether tethered peptides. The time dependency of dealkylation process was also studied by CD. The CD spectra of peptide 5b displayed α-helix at the beginning then gradually switched to a random coil conformation as shown in Fig. S13 in Supporting information.
To test whether this concept could be translated into longer peptides, model peptide FITC-βA-(cyclo-5, 9)-HKILmS5RLLCDSNH2 (11) targeting estrogen receptor (ER-α) [19] and its 4-bromomethyl-benzyl octanoate alkylated analogue (12) were synthesized [20], and peptide 12 shows enhanced helical content and cell permeability compared with peptide 11 (Fig. S14 in Supporting information).
In summary, we have developed an in-tether chiral sulfonium strategy for reversibly switching peptide conformation by alkylation and dealkylation of the sulfonium auxiliary under mild conditions. We could not get the crystal structure of sulfonium peptides to determine the absolute configuration of the sulfonium center. However, we previously reported a carbon or a sulfoxide chiral center at the same position both need to be R for helicity inducement [3d, 6a], thus, we believe the sulfonium center will prefer the same absolute configuration. This strategy broadened our scope of CIH strategy and provided a valuable approach to functionalize the peptide tether. The release of the parent unmodified peptides is unique and not seen in previously reported on-tether modification of constraint peptides [21]. This traceless redox sensitive tagging strategy could be utilized for various biological application, such as fabrication of detachable cell penetrating peptides for cargo delivery [22]. Further investigations and biological applications of this sulfonium auxiliary is under current investigation in the laboratory and will be reported in due course.
AcknowledgmentsWe acknowledge financial support from the National Natural Science Foundation of China (Nos. 21372023 and 81572198); Ministry of Science and Technology of the People's Republic of China (No. 2015DFA31590); the Shenzhen Science and Technology Innovation Committee (Nos. JSGG20140519105550503, JCYJ20150331100849958, JCYJ20150403101146313, JCYJ20160301111338144, JCYJ20160331115853521, JSGG20160301095829250 and ZDSYS201504301539161); the Shenzhen Peacock Program (No. KQTD201103).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.07.003.
| [1] |
(a) A. P. Higueruelo, H. Jubb, T. L. Blundell, Curr. Opin. Pharmacol. 13(2013) 791-796; (b) M. Pelay-Gimeno, A. Glas, O. Koch, T. N. Grossmann, Angew. Chem. Int. Ed. 127(2015) 9022-9054. |
| [2] |
H. Yin, A.D. Hamilton, Angew. Chem. Int. Ed. 44 (2005) 4130-4163. DOI:10.1002/(ISSN)1521-3773 |
| [3] |
(a) C. E. Schafmeister, J. Po, G. L. Verdine, J. Am. Chem. Soc. 122(2000) 5891-5892; (b) R. N. Chapman, G. Dimartino, P. S. Arora, J. Am. Chem. Soc. 126(2004) 12252-12253; (c) N. E. Shepherd, H. N. Hoang, G. Abbenante, D. P. Fairlie, J. Am. Chem. Soc. 127(2005) 2974-2983; (d) K. Hu, H. Geng, Q. Zhang, et al., Angew. Chem. Int. Ed. 55(2016) 8013-8017; (e) Y. Jiang, K. Hu, X. Shi, et al., Org. Biomol. Chem. 15(2017) 541-544; (f) H. Lin, Y. Jiang, K. Hu, et al., Org. Biomol. Chem. 14(2016) 9993-9999; (g) Y. Tian, J. Li, H. Zhao, et al., Chem. Sci. 7(2016) 3325-3330; (h) H. Zhao, Q. S. Liu, H. Geng, et al., Angew. Chem. Int. Ed. 128(2016) 12267-12272; (i) O. Khakshoor, J. Nowick, Curr. Opin. Chem. Biol. 12(2008) 722-729; (j) D. Y. Jackson, D. S. King, J. Chmielewski, S. Singh, P. G. Schultz, J. Am. Chem. Soc. 113(1991) 9391-9392; (k) H. Jo, N. Meinhardt, Y. Wu, J. Am, et al., Chem. Soc. 134(2012) 17704-17713; (l) A. M. Spokoyny, Y. Zou, J. J. Ling, et al., J. Am. Chem. Soc. 135(2013) 5946-5949; (m) S. Samanta, C. Qin, A. J. Lough, G. A. Woolley, Angew. Chem. Int. Ed. 51(2012) 6452-6455; (n) H. K. Cui, Y. Guo, Y. He, et al., Angew. Chem. Int. Ed. 52(2013) 9558-9562; (o) Y. Guo, D. M. Sun, F. L. Wang, et al., Angew. Chem. Int. Ed. 54(2015) 14276-14281. |
| [4] |
B.N. Bullock, A.L. Jochim, P.S. Arora, J. Am. Chem. Soc. 133 (2011) 14220-14223. DOI:10.1021/ja206074j |
| [5] |
H. Lin, Y. Jiang, Q. Zhang, K. Hu, Z. Li, Chem. Commun. 52 (2016) 10389-10391. DOI:10.1039/C6CC04508A |
| [6] |
Q. Zhang, F. Jiang, B. Zhao, et al., Sci. Rep. 6 (2016) 38573. DOI:10.1038/srep38573 |
| [7] |
(a) J. Li, Y. Tian, D. Wang, et al., Bioorg. Med. Chem. 25(2017) 1756-1761; (b) S. L. Schreiber, Science 287(2000) 1964-1969; (c) A. K. Yudin, Chem. Sci. 6(2015) 30-49; (d) N. Assem, D. J. Ferreira, D. W. Wolan, P. E. Dawson, Angew. Chem. Int. Ed. 54(2015) 8665-8668; (e) Y. H. Lau, P. de Andrade, S. T. Quah, et al., Chem. Sci. 5(2014) 1804-1809. |
| [8] |
(a) J. A. Johnson, Y. Y. Lu, J. A. Van Deventer, D. A. Tirrell, Curr. Opin. Chem. Biol. 14(2010) 774-780; (b) S. Lin, X. Yang, S. Jia, et al., Science 355(2017) 597-602. |
| [9] |
J.R. Kramer, T.J. Deming, Chem. Commun. 49 (2013) 5144-5146. DOI:10.1039/c3cc42214c |
| [10] |
(a) T. J. Deming, Bioconjugate Chem. 28(2017) 691-700; (b) T. J. Deming, Chem. Rev. 116(2016) 786-808. |
| [11] |
(a) B. Zhao, Q. Zhang, Z. Li, J. Pept. Sci. 22(2016) 540-544; (b) A. D. de Araujo, H. N. Hoang, W. M. Kok, et al., Angew. Chem. Int. Ed. 53(2014) 6965-6969. |
| [12] |
Y. Zhang, M.P. Hudock, K. Krysiak, et al., J. Med. Chem. 50 (2007) 6067-6079. DOI:10.1021/jm700991k |
| [13] |
Q. Zhang, X. Shi, Y. Jiang, Z. Li, Tetrahedron 70 (2014) 7621-7626. DOI:10.1016/j.tet.2014.08.004 |
| [14] |
(a) P. C. Lyu, M. I. Liff, L. A. Marky, N. R. Kallenbach, Science 250(1990) 669-673; (b) Y. Demizu, M. Doi, M. Kurihara, et al., Org. Biomol. Chem. 9(2011) 3303-3312; (c) J. Sola, M. Bolte, I. Alfonso, Org. Biomol. Chem. 13(2015) 10797-10801. |
| [15] |
N.J. Greenfield, Nat. Protoc. 1 (2006) 2876-2890. |
| [16] |
A. Pardi, M. Billeter, K. Wuthrich, J. Mol. Biol. 180 (1984) 741-751. DOI:10.1016/0022-2836(84)90035-4 |
| [17] |
J. Graf, P.H. Nguyen, G. Stock, H. Schwalbe, J. Am. Chem. Soc. 129 (2007) 1179-1189. DOI:10.1021/ja0660406 |
| [18] |
A.D. Araujo, H.N. Hoang, W.M. Kok, et al., Angew. Chem. Int. Ed. 53 (2014) 6965-6969. DOI:10.1002/anie.201310245 |
| [19] |
(a) C. Phillips, L. Roberts, M. Schade, et al., J. Am. Chem. Soc. 133(2011) 9696-9699; (b) S. Fuchs, H. D. Nguyen, T. T. Phan, et al., J. Am. Chem. Soc. 135(2013) 4364-4371. |
| [20] |
G.K. Dewkar, P.B. Carneiro, M.C. Hartman, Org. Lett. 11 (2009) 4708-4711. DOI:10.1021/ol901662c |
| [21] |
Y. Zou, A.M. Spokoyny, C. Zhang, et al., Org. Biomol. Chem. 12 (2014) 566-573. DOI:10.1039/C3OB42168F |
| [22] |
Z. Qian, X. Xu, J.F. Amacher, et al., Angew. Chem. Int. Ed. 54 (2015) 5874-5878. DOI:10.1002/anie.201411594 |