 2018, Vol. 29
2018, Vol. 29 



Recently, the vicinal difunctionalization of alkenes has been increasing in importance for rapidly increasing molecular complexity with a variety of functional groups [1-5]. The hydroxyselenenylation of alkenes is a type of difunctionalization of alkenes, of which, both a selenium atom and hydroxy group can be installed into the carbon–carbon double bond. Due to β-Hydroxy selenide selenides are valuable intermediates in the synthesis of allylic alcohols [6], olefins [7], bromohydrins [8] vinyl selenides [9, 10], and some important natural compounds [11-13], several methods are available for their preparation. The electrophilic addition of the commercially available selenenylating reagent PhSeCl to alkenes is a useful procedure [14, 15]; however, the presence of toxic and moisture-sensitive nature of it, and the nucleophilic Cl- anion can give rise to undesirable side processes. Alternatively, some novel reagents which do not contain nucleophilic counterions, such as PhSeOSO2Ar, N-phenylselenophtalimide and N-phenylselenosuccinimide, have been developed [16-18]. A simpler method for formation of the electrophilic phenylselenium cation is oxidation of less expensive and less toxic diphenyl diselenide with oxidants, like DDQ, iodobenzene diacetate or electrolytic system [19-21]. Using selenolate anions, the SN2 ring-opening of epoxides is another way for access to β-Hydroxy selenide selenides. The selenolate anions can be generated by treatment diphenyl diselenide with sodium, zinc, sodium borohydride, zinc/aluminium (Ⅲ) chloride, tributylphosphine in an alkaline medium and sodium hydroxymethanesulfinate [22-27]. More recently, Wu and coworkers reported a new copper-catalyzed cross-coupling of aryl iodides, epoxides, and elemental selenium for synthesis of β-Hydroxy selenide selenides [28]. However, these in situ generated selenolate anions are relatively unstable. They should be prepared under controlled anhydrous conditions and some of above methods require higher temperature or longer reaction time, which restrict their applications. Therefore, it is of significant interest to find mild and simple methods for the synthesis of β-Hydroxy selenide selenides.
Over the past few years, iodine-catalyzed or iodine-mediated reactions have been increasingly explored because iodine is cheap, readily available and eco-friendly, especially it has the metal-like behavior [29-33]. In our recent research, we found that some inorganic haloid salts combined with suitable oxidants or molecular iodine can promote the oxidative cleavage of the Se– Se bond of diphenyl diselenide, and some new electrophilic reactions have been reported [34-37]. Based on this, we investigated the reaction of alkenes with diselenides in the presence of molecular iodine. Herein, we wish to report a novel and simpler regioselective hydroxyselenenylation of alkenes mediated by I2. To the best of our knowledge, this method has not been reported before.
The typical procedure for the I2-mediated hydroxyselenenylation of alkenes: alkene 1a (0.24 mmol), diselenide 2a (0.10 mmol) and I2 (0.10 mmol) were added successively to MeCN/H2O (3 mL, 1/1, v/v). The mixture was vigorously stirred at room temperature for 12 h. Upon completion, the reaction was quenched by addition of sat. aq. Na2S2O3 (2 mL), then sat. aq. Na2CO3 (2 mL) and H2O (5 mL) were added, respectively. The mixture was extracted with CH2Cl2 (5 mL × 3) and the combined organic phase was dried over anhydrous Na2SO4, then filtered and concentrated under reduced pressure. The residue was finally purified on a silica gel plate (petroleum ether/ethyl acetate, 4:1) to furnish product 3a as pale yellow oil [18] in 87% yield. 1H NMR (500 MHz, CDCl3): δ 7.56 (dd, 2H, J = 6.4, 2.8 Hz), 7.45-7.17 (m, 8H), 4.77 (dd, 1H, J = 9.4, 3.6 Hz), 3.32 (dd, 1H, J = 12.8, 3.7 Hz), 3.16 (dd, 1H, J = 12.8, 9.4 Hz); 13C NMR (125 MHz, CDCl3): δ 142.5, 133.1, 129.24, 129.20, 128.5, 127.9, 127.4, 125.8, 72.3, 38.4; IR (film, cm-1): ν 3500–3100, 3059, 3030, 1477, 1437, 1054, 1023, 737, 700; MS (EI, m/z, %): 295 (M + 18, 42).
Firstly, to develop simpler methods for the synthesis of β-Hydroxy selenide selenides, we investigated the reaction of styrene 1a with diphenyl diselenide 2a in the presence of I2 at room temperature. It was found that on simple stirring of the mixture of 1.2 equiv. of 1a, 1.0 equiv. of 2a and 1.0 equiv. of I2 in H2O for 24 h at room temperature, the expected hydroxyselenenylation of styrene was not carried out, and the substrates 1a and 2a were recovered in near quantitatively (Table 1, entry 1). Fortunately, when a mixed solvent MeCN/H2O (5/1, v/v) took the place of H2O, the same reaction proceeded smoothly, and after 24 h the desired addition product, 1-phenyl-2-(phenylselanyl)ethanol 3a, was obtained in 69% yield (entry 2). Then, the hydroxyselenenylation of 1.2 equiv. of 1a with 1.0 equiv. of 2a mediated by I2 at room temperature was optimized. It is obvious that the reaction was completed in 12 h (entries 2-4). Compared with MeCN/H2O (5/1), other two mixed solvents of CH2Cl2/H2O and EtOAc/H2O were evaluated, and in which EtOAc/H2O had the best effect (entries 3, 5, 6). Following, the suitable volume ratio of EtOAc to H2O was examined, and when it reached 1:1, the best yield of 87% was obtained (entries 6–9). Finally, the optimal amount of I2 was screed and 1.0 equiv. proved to be the best choice (entries 8, 10, 11). However, in the absent of I2, 3a was not determined (entry 12).
|
|
Table 1 Optimization of the hydroxyselenenylation of styrene mediated by I2. |
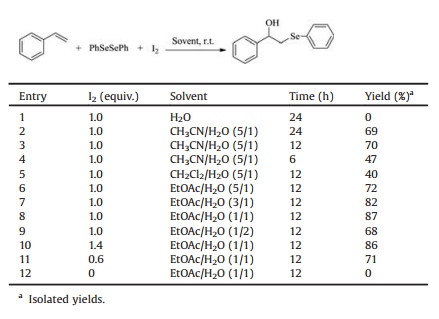
After extensive screening of reaction conditions, we concluded that the most efficient set of conditions employs 1.2 equiv. of alkenes 1, 1.0 equiv. of diselenides 2 and 1.0 equiv. of I2 in EtOAc/ H2O (1/1) at room temperature for 12 h. On the basis of these screening results, to demonstrate the scope of this reaction, a detailed study was performed with various terminal alkenes and diselenides. The results are summarized in Table 2.
|
|
Table 2 Preparation of β-Hydroxy selenide selenides 3. |

As shown in Table 2, the I2-mediated hydroxyselenenylation was compatible with the studied alkenes 1a-1h, affording the corresponding β-Hydroxy selenide selenides 3a–3h in good to excellent yields (Table 2, entries 1-8). It is obvious that the substituents on benzene ring for these aromatic alkenes, whether they were electron-donating (Me, AcO and t-Bu) or electron-withdrawing (Cl, Br and NO2) groups, usually had no significant influence on their reactivity (entries 2-7). Similar to 1a, 1h also provided the corresponding product 3h in near same yield, which means that the methyl group in 1h had not the steric effect (entries 1, 8). However, when 2-vinylpyridine was treated under the same conditions, the desired product was obtained in only 22% yield. When cyclohexene 1i and 1-hexene 1j were treated as the representatives of aliphatic alkenes, somewhat low yields of 71% and 70% were obtained for 3i and 3j, respectively, indicating our protocol is more suitable for aromatic alkenes. Dibenzyl diselenide 2b, similar to 2a, an aliphatic diselenide was also effective in the reaction, resulting in the corresponding products 3k–3n in good yields (entries 11–14). All the data and spectra have been deposited in Supporting information.
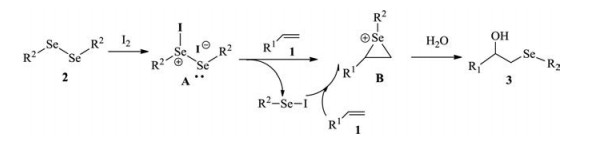
According to the above mentioned results and control experiment, a plausible eletrophilic addition mechanism mediated by I2 is shown in Scheme 1. Thus, molecular I2 first reacts smoothly with diselenide 2 to form the active intermediate A, followed by a rapid cleavage of Se-Se bond [34, 38]. The in situ generated active electrophilic selenium species then reacts with alkene 1 to produce the unstable cyclic seleniranium intermediate B. Finally, intermediate B is attacked by water to provide the desired product 3 as a single isomer via an SN1 mechanism. When aliphatic alkene 1i is treated in the reaction, the trans stereoisomer via an SN2 mechanism is obtained [20]. Accompanying the reaction, another active intermediate ArSeI [39] can further transfer a second equivalent of electrophilic selenium to alkene 1.

|
Download:
|
| Scheme 1. Proposed mechanism for the hydroxyselenenylation of alkenes mediated by I2. | |
{kind=link}
In summary, we developed a new and convenient procedure for the regioselective preparation of β-Hydroxy selenide selenides from alkenes, diselenides and I2 at room temperature. This iodinemediated hydroxyselenenylation of alkenes has some advantages such as mild reaction conditions and simple procedure, which provides a series of β-Hydroxy selenide selenides with high regioselectivity and in good yields. Furthermore, this reaction will extend the application scope of molecular iodine in organic synthesis.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.06.023.
| [1] |
R.M. Romero, T.H. Woste, K. Muniz, Chem.-Asian J. 9 (2014) 972-983. DOI:10.1002/asia.v9.4 |
| [2] |
E.M. Beccalli, G. Broggini, S. Gazzola, et al., Org. Biomol. Chem. 12 (2014) 6767-6789. DOI:10.1039/C4OB00610K |
| [3] |
B. Zhao, X. Peng, Y. Zhu, et al., J. Am. Chem. Soc. 133 (2011) 20890-20900. |
| [4] |
H.W. Zhang, Y.C. Song, J.B. Zhao, et al., Int. Ed. 53 (2014) 11079-11083. DOI:10.1002/anie.201406797 |
| [5] |
C. Chen, M.B. Hecht, A. Kavara, et al., J. Am. Chem. Soc. 137 (2015) 13244-13247. DOI:10.1021/jacs.5b08611 |
| [6] |
K.B. Sharpless, R.F. Lauer, J. Am. Chem. Soc. 95 (1973) 2697-2699. DOI:10.1021/ja00789a055 |
| [7] |
H.J. Reich, F. Chow, J. Chem. Soc., Chem. Commun. 3 (1975) 790-791. |
| [8] |
M. Sevring, W. Dumont, L. Hevesi, et al., Tetrahedron Lett. 17 (1976) 2647-2650. DOI:10.1016/S0040-4039(00)91758-1 |
| [9] |
D. Van Ende, W. Dumont, A. Krief, Angew Chem. Int. Ed.Ed. Engl.(1975), 700-702. |
| [10] |
W. Dumont, A. Krief, Angew. Chem. Int. Ed. Engl.(1975), 350-351. |
| [11] |
J.H. Rigby, U.S.M. Maharoof, M.E. Mateo, J. Am. Chem. Soc. 122 (2000) 6624-6628. DOI:10.1021/ja000930i |
| [12] |
E.M. Treadwell, J.D. Neighbors, D.F. Wiemer, Org. Lett. 4 (2002) 3639-3642. DOI:10.1021/ol0266368 |
| [13] |
S. Knapp, D.L. Zhao, Org. Lett. 2 (2000) 4037-4040. DOI:10.1021/ol0066680 |
| [14] |
A. Toshimitsu, T. Aoai, H. Owada, et al., Tetrahedron 41 (1985) 5301-5306. DOI:10.1016/S0040-4020(01)96781-X |
| [15] |
A. Toshimitsu, T. Aoai, H. Owada, et al., J. Chem. Soc., Chem. Commun.(1980), 412-413. |
| [16] |
M. Tingoli, R. Diana, B. Panunz, Tetrahedron Lett. 47 (2006) 7529-7531. DOI:10.1016/j.tetlet.2006.08.068 |
| [17] |
K.C. Nicolaou, D.A. Claremon, W.E. Barnette, et al., J. Am. Chem. Soc. 101 (1979) 3704-3706. DOI:10.1021/ja00507a069 |
| [18] |
M. Yoshshida, S. Sasage, K. Kawamura, et al., Bull. Chem. Soc. Jpn. 64 (1991) 416-422. DOI:10.1246/bcsj.64.416 |
| [19] |
M. iecco, L. Testaferri, A. Temperini, Synlett(2001), 1767-1771. |
| [20] |
M. Tingoli, M. Tiecco, L. Testaferri, et al., Synth. Commun. 28 (1998) 1769-1778. DOI:10.1080/00397919808007007 |
| [21] |
S. Torii, K. Uneyama, M. Ono, Tetrahedron Lett. 21 (1980) 2741-2744. DOI:10.1016/S0040-4039(00)78594-7 |
| [22] |
S.V. Ley, L.A. O'Neil, C.M.R. Low, Tetrahedron 42 (1986) 5363-5368. DOI:10.1016/S0040-4020(01)82086-X |
| [23] |
M. Barahman, S. Mojgan, Synlett(2005), 1316-1318. |
| [24] |
S. Berlin, C. Ericsson, L. Engman, J. Org. Chem. 68 (2003) 8386-8396. DOI:10.1021/jo030153f |
| [25] |
B. Movassagh, S. Farshbaf, Synthesis(2010), 33-35. |
| [26] |
M. Sakakibara, K. Katsumata, Y. Watanabe, et al., Synthesis(1992), 377-379. |
| [27] |
V. Ganesh, C. Srinivasan, Synthesis(2009), 3267-3278. |
| [28] |
L. Min, G. Wu, M.C. Liu, et al., J. Org. Chem 81 (2016) 7584-7590. DOI:10.1021/acs.joc.6b01274 |
| [29] |
R.D. Richardson, T. Wirth, Angew Chem. Int. 45 (2006) 4402-4404. DOI:10.1002/(ISSN)1521-3773 |
| [30] |
J. Feng, S. Liang, S.Y. Chen, et al., Adv. Synth. Catal. 354 (2012) 1287-1292. DOI:10.1002/adsc.201100920 |
| [31] |
K. Yang, M. Ke, Y.G. Lin, Green Chem. 17 (2015) 1395-1399. DOI:10.1039/C4GC02236J |
| [32] |
Y.N. Duan, Z. Zhang, C. Zhang, Org. Lett. 18 (2016) 6176-6179. DOI:10.1021/acs.orglett.6b03209 |
| [33] |
S. Ambethkar, M. Vellimalai, V. Padmini, et al., New J. Chem. 41 (2017) 75-80. DOI:10.1039/C6NJ02102F |
| [34] |
H.W. Shi, C. Yu, M. Zhu, et al., J. Organomet. Chem. 776 (2015) 117-122. DOI:10.1016/j.jorganchem.2014.10.025 |
| [35] |
H.W. Shi, C. Yu, J. Yan, Chin. Chem. Lett. 26 (2015) 1117-1120. DOI:10.1016/j.cclet.2015.05.029 |
| [36] |
C. Yu, H.W. Shi, M. Zhu, et al., Synlett 26 (2015) 2139-2144. DOI:10.1055/s-00000083 |
| [37] |
Y.K. Zhang, S.X. Wu, J. Yan, Helv Chim. Acta 99 (2016) 654-658. DOI:10.1002/hlca.201600115 |
| [38] |
C. Muangkaew, P. Katrun, P. Kanchanarugee, et al., Tetrahedron 69 (2013) 8847-8856. DOI:10.1016/j.tet.2013.08.018 |
| [39] |
Z.Z. Huang, X. Huang, Y.Z. Huang, J. Chem. Soc., Perkin Trans. I(1995), 95-96. |