 2018, Vol. 29
2018, Vol. 29 



Organic semiconductors have attracted great interests in relation to their utilities in optoelectronics and molecular electronics [1]. In contrast to rich varieties of p-channel organic semiconductor [2], n-channel organic semiconductor are limited to certain electron deficient compounds [3, 4] and still have much room for improvement in mobility, stability, and processability [5]. In this context, tetracarboxylic diimides [6-9] are important category since they can tune HOMO-LUMO band gap by using different conjugated backbones and allow solution processing while modified at diimide termini. So far, naphtalene-[6], anthracene-[7], perylene-[8], terrylene-[9], and dibenzotetrathiafulvalene-based diimides [10] have been synthesized and the best of them shows medium carrier mobility at 0.01 cm2 V-1 s-1 order [8]. On the other hand, phenazine derivatives due to the presence of electron deficient imine moieties, are another promising candidates as suggested by the results of cyclic voltammetry measurements [11] and theoretical calculations [12], although their transportation properties are yet to be proved experimentally. In contrast to acenes, one appealing characteristic of phenazines is their high stability against thermal decomposition and photobleaching [13]. Considering the virtues of phenazine and tetracarboxylic imides, we were motivated to explore the possibility to integrate these components chemically and deduced that the combination of phenazine pigment with diimide functionality would substantially enhance electron affinity and thermal stability while keeping "flexibility" for structure modification at both the focal core and the diimide termini. Herein, we report a novel organic n-type semiconductor, i.e., pyrenephenazine tetracarboxylic diimide and monoimide (Fig. 1, PPDI and PPMI), which allows for the first time an elaborate fusion of the above structure parameters. PPDI was synthesized by condensation of 1, 2-diamino-4, 5-phthalimide with pyrene-4, 5, 9, 10-tetranone in an HOAc/EtOH mixture under reflux condition and unambiguously characterized by spectroscopies (Supporting information).

|
Download:
|
| Fig. 1. Molecular structures of PPDIs 1, and PPMIs 2. | |
General methods are explained in Section 1 in Supporting information. Unless otherwise stated, reagents were commericially obtained and used without further purification. Pyrene-4, 5, 9, 10-tetraketone 10 and pyrene-4, 5-dione 11 were prepared according to reported methods [18]. PPDI 1a were synthesized by condensation of diamino-substituted phthalimide with pyrene-4, 5, 9, 10-tetranone in HOAc/EtOH mixture under reflux condition (Scheme 1 and Section 2 in Supporting information). In a typical procedure, the resulted yellow precipitation of the reaction mixture was collected by filtration and the solid was rinsed with EtOH, to afford 1a in a good yield of 71%. MALDI-TOF mass spectra revealed a single species in good accordance with the calculations (Fig. S1 in Supporting information). The strong π-π stacking and the rigid structure made 1a almost insoluble in common organic solvents at room temperature, making routine characterization (1H NMR, 13C NMR) impossible. To obtain a soluble analogue, dodecyl alphatic chain was introduced to the imide terminal at the earlier diamino-phthalimide step. Further condensation with pyrene-4, 5, 9, 10-tetranone under the same condition give the dodecylated PPDI 1b in 93% yield. PPMIs 2 were easily synthesized by condensation with pyrene-4, 5-dione 11 instead of 10 accordingly in a relative high yields (Scheme 1 and Section 2 in Supporting information).

|
Download:
|
| Scheme 1. Synthesis of PPDIs 1 and PPMIs 2. | |
1a is hardly soluble in common organic solvents such as THF, dichloromethane, chloroform, acetone etc., while slightly soluble in 1, 2-dichlorobenzene upon heating. Electronic absorption spectroscopy of 1a in 1, 2-dichlorobenzene at 100 ℃ displays peaks at 330, 386, 408, and 432 nm (Fig. 2a, black line), which is about 97-nm red-shifted in comparison with that of pyrene (335 nm, in THF) [12]. This large red-shift suggests the π-conjugation of 1a is extended over the molecular skeleton. Molecular modeling optimized at the AM1 level reveals that 1a is a planar molecule, where the focal pyrene unit and bisimide terminals are in the same plane without any twist or distortion. The rigid molecule adopts a tape-like structure with a length of about 21 Å and a width of about 9 Å, respectively. In contrast, the dodecyl chain tailored bisimide 1b show higher solubility (2.3 mmol/L in THF) than 1a (0.46 mmol/L in 1, 2-dichlorobenzene). However, 1b was also found to be easily aggregated to form much softer nanobelts (Fig. 3b) due to the large π-conjugated core and van der Waals interaction of the long alphatic chains. On the other hand, monoimides 2a and 2b show much higher solubility and stronger fluorescence comparing with their analogues bisimides 1a and 1b. For example, 2a was soluble in DMSO (2.7 mmol/L) upon slightly heating; 2b was highly soluble in common organic solvents like CH2Cl2, CHCl3, THF and acetone at room temperature without aggregation. It is also found that these two asymmetric planar molecules are very easy to form nanobelts using a binary solvents exchanging self-assembly procedure (Section 5 in Supporting information) [13]. Both bisimide 1a and 1b emit blue emission at 441 nm while the monoimide 2a and 2b exhibit a much stronger green emission at 538 nm and 510 nm, respectively (Figs. 2c and d). The difference of the maxim absorption and emission peaks between 2a and 2b may due to the solvent effect. It is noteworthy that all the excitation spectra of these four compounds from the longest emission overlap with their absorption spectra very well. Moreover, bisimide 1 exhibit much smaller stoke shift (9 nm and 12 nm in the case of 1a and 1b, respectively) compared with the monoimide 2 (84 nm and 48 nm for 2a and 2b, respectively). These may ascribe to the symmetric and rigid structure in the case of bisimide. The energy loss would be much smaller compared with those of monoimide when irradiated under UV light.

|
Download:
|
| Fig. 2. UV–vis absorption (black line), fluoroescence emission (blue or green line) and excitation spectra (red line) of PPIs. a) 1a (λex = 331 nm, λem = 441 nm); b) 1b (λex = 330 nm, λem = 442 nm); c) 2a (λex = 347 nm, λem = 538 nm); d) 2b (λex = 346 nm, λem = 510 nm). | |

|
Download:
|
| Fig. 3. FE-SEM images of self-aseembled PPIs. a) 1a; b) 1b; c) 2a; d) 2b. | |
To investigate the self-assembly behaviour, the as-synthesized samples were dispersed in different solvents and heated to reflux. After cooling to room temperature, the morphologies of the precipitates were investigated by Field emission scanning electron microscopy (FE-SEM, Fig. 3). We found that 1a exhibited much better solubility in DMSO like its phthalmide precursors 4–9 (all dissolved in DMSO for NMR measurement). In a typical selfassembly process, PPDI 1a was suspended in a mixture of 1, 2-dicholorobenzene/DMSO (1:1, v/v) and gives a clear solution (0.25 mg/mL) upon refluxing, which was then cooled at 25 ℃ to afford yellow precipitates. The yellow precipitates were collect by centrifugation and washed with ethanol and dried under vacuum. FE-SEM images of the precipitate (re-dispersed in ethanol) thus obtained reveals one dimensional straight tape structures (Fig. 3a), indicating that PPDI 1a self-assembled in the above condition. The self-assembled tapes have a length of about tens of micrometers a length of about tens of micrometers and width of about 100– 300 nm (Fig. 3a, Fig. S4 in Supporting information). AFM images (in tapping mode) display that the typical height of the tapes is about 50 nm (Fig. S4). The transmission electron microscopy (TEM) images are consistent with the SEM ones, and the selected-area electron diffraction pattern, which observed from a single belt revealed that each belt had a polycrystalline structure (Fig. S3 inset in Supporting information). Powder X-ray diffraction pattern exhibited two primary sharp reflections at 2θ = 6.84°, 12.60° corresponding to d spacing of 12.91 Å, 7.02 Å. Several weak but sharp scattering was observed at 8.40°, 16.86°, 19.42°, 20.74° (Fig. S5 in Supporting information), and 26.60°. The typical π-π stacking distance was observed to be 3.35 Å. These features are typical of 1D nano-architectures assembled from planar π-conjugated organic molecules and suggest that PPIs self-assembled in a fashion of interdigitated dimers, which further π-π stacked generate the tape structure. The other three imides all formed nanobelts upon tuning proper assembly conditions. Bright spot in SAED pattern and sharp reflection peaks from TEM and PXRD measurements confirmed that all these PPIs assembled to form highly crystalline nanobelts.
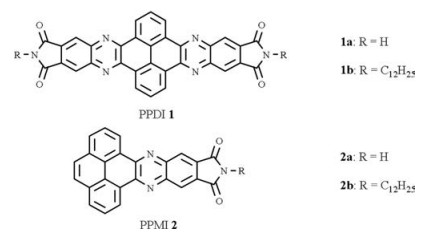
Due to their low solubility, thin film cyclic voltammetry was carried out to characterize the redox potential of nonalkylated PPIs 1a and 2a in dichloromethane using ITO glasses as the counter and working electrodes. The assembled nanobelt suspension in dichloromethane was spin-coated on ITO glass serving as the working electrodes. The dodecylated imides 1b and 2b were measured by the conventional method in CHCl3 solution (0.5 mmol/L). As shown in Fig. 4, bisimides 1a, 1b showed almost same reversible pair at -0.93/-0.81 V and -0.96/-0.83 vs. Fc+/Fc. Combined with electronic absorption spectrum, the HOMO and LUMO are estimated to be -6.57 eV and -3.89 eV, respectively for 1a. In sharp contrast, the monoimides 2a and 2b show almost identical CV profiles with two reversible voltammetric waves at -1.64 V, -1.36 V and a quasi-reversible peak about -1.20 V. These results indicate that all PPIs are n-type chromophore.

|
Download:
|
| Fig. 4. Cyclic voltammograms for the reduction of PPIs. a) 1a; b) 1b; c) 2a; d) 2b. | |
To further evaluate the electronic properties of these four compounds, density functional theory (DFT) calculations were performed using Gaussian 03 at the B3LYP/6-31G(d, p) level. The theoretically predicted HOMO-LUMO orbitals for all four compounds are presented in Fig. S7 in Supporting information. All the energy values from electrochemistry and theoretical calculation are summarized in Table 1 for comparison. The experimental values of HOMO-LUMO energies are in reasonable agreement with the calculated ones. The two reversible voltammetric waves observed in monoimide 2 indicate a two steps one electron reduction process due to the asymmetric structure. However, the bisimide 1 only displayed one reversible pair reduction peaks, indicating that all pyrazine rings are identical due to the highly symmetric structure. The quasi-reversible reduction potential at -1.20 V observed in monomide 2a and 2b may refer to the reduction of the unfused pyrene edge. As shown in Fig. S7, the largest coefficients in the HOMO orbital are mainly located on the central pyrene core. As expected, the coefficients in the lowest unoccupied molecular orbital (LUMO) orbital are mostly located on two phenazine and two imide rings. The results indicate that there may be intramolecular charge transfer. Thermal gravity analysis under nitrogen upon heating from 25 ℃ to 800 ℃ at a rate of 10 ℃/min shows that PPI 1a is extremely stable without any decompose below 470 ℃ and only shows slight weight loss of about 23% upon heating at 600 ℃ (Fig. S6 in Supporting information).
|
|
Table 1 Summary of electronic properties of PPIs 1-2 from experiments and calculation. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Since PPI molecules easily form well-defined nanobelts, we expect that this molecular alignment should benefit the carrier transfer along these ordered nanostructures. The electron conductivity was measured on a Keithley model 6430 subfemtoampere sourcemeter by a two probes method. The I–V curve of a drop-cast film on a 10 μm gap Pt conductive electrode on ITO surface was shown in Fig. 5. The bare sample of 1a show low conductivity with the current range of -2 nA to 0.3 nA with a -2V ~ 2 V bias. In contrast, the same sample dopping with triethylamine vapour exhibit much large current around -50 nA to 35 nA. In the case of 2a, similar results was observed. The Et3N dopped nanotapes of 2a show much large current around -760 pA to 470 pA, which is about 150 times larger than the bare sample with the current range of -5 pA to 3 pA at a 2 V bias. As for the dodecylated analogues 1b and 2b, each show slightly increased current at the same magnitude order with its nontailored analogues. These may due to the van der Waals interaction between the alphatic chains facilitated the molecular packing to form more ordered structure. Triethylamine can also be severed as the dopant for 1b and 2b with a much increased current (Figs. 5b and d, blue curve). These results indicate an electron transfer from the electron donor (Et3N) to the acceptor (PPIs) [14] which further confirm that PPIs is an n-channel semiconductor.

|
Download:
|
| Fig. 5. Current (I)–voltage (V) profiles at 25 ℃ of cast bare films (red line) and films dopping with triethylamine vapour (blue line) of PPIs on a 10 micrometer gap Pt electrode imbed on glass substrate. a) 1a; b) 1b; c) 2a; d) 2b. | |
{kind=link}
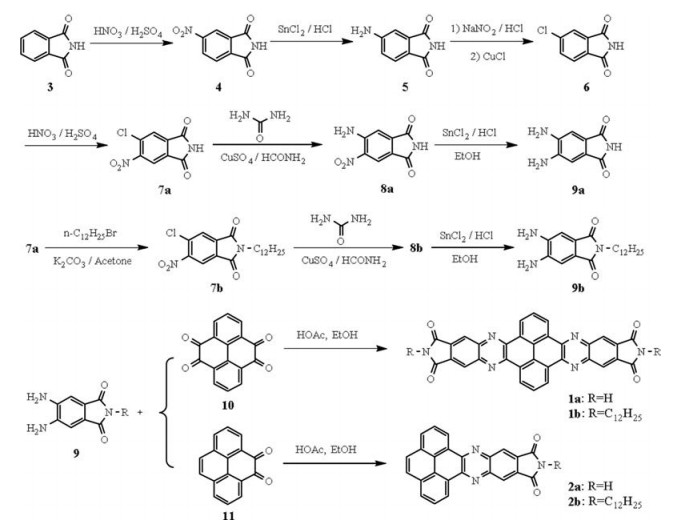
We utilized laser flash photolysis time-resolved microwave conductivity (FP-TRMC) technique to investigate the intrinsic carrier mobility [15]. Transient conductivity measurement displays a rapid rise upon laser irradiation at 355 nm in both cases. The ΦΣμmax values observed for bisimide 1 were in the range of 0.66 × 10-4–1.36 × 10-4 cm2 V-1 s-1 in Ar gas at the photon density of 4.5 × 1016 photons cm-2 (Figs. 6a and b). On the other hand, the two monoimides 2 show much lower mobility with Σμmax values range from 2.87 × 10-6 cm2 V-1 s-1 to 3.60 × 10-6 cm2 V-1 s-1. In order to determine the number of charge carriers, time-of-flight transient integration was measured at different bias voltages. The number of charge carrier at 2 V was observed to be 7.1 × 1011, thus the charge carrier generation yield (Φ = number of charge carrier/ photon) was evaluated to be 1.58 × 10-5. The minimum carrier mobility (Σμ) of 1a was thus calculated to be 4.14 cm2 V-1 s-1, which is much larger compared with the similar systems [16]. The carrier mobility of the other three imides 1b, 2a, and 2b were calculated to be 0.57, 0.23 and 0.18 cm2 V-1 s-1, respectively. It is obvious that the mobility of bismides 1 is much larger than monoimides 2 due to the larger π-conjugation and efficient π-π stacking which favours the charge transfer through the assembled columns. On the other hand, the carrier mobility of both the alkylated bisimide 1b and monoimide 2b is about ten times smaller than their non-alkylated analogues 1a and 2a.

|
Download:
|
| Fig. 6. FP-TRMC profiles at 25 ℃ of PPIs. a) 1a; b) 1b; c) 2a; d) 2b. | |
{kind=link}
It has been reported that pyrazine and phenazine can be protonated by addition of strong acid [17]. We found that an obvious color change and fluorescence quench upon addition of TFA to the solution of bisimide 1b and monoimide 2b in CHCl3 (0.01 mg/mL). There was a significant color change from yellow to blue upon the addition of TFA to 2b solution in CHCl3. This result implied that these soluble imides could be a potential colorimetric acid sensor. The TFA titration experiments were monitored by UVvis and fluorescence spectroscopy. The absorbances at 332, 347, 442 and 462 nm decreased gradually while new absorption at 356 and 590 nm became dominant as the amount of TFA increased (Fig. 7c). Meanwhile, the original green emission at 510 nm was gradually quenched by addition of TFA (Fig. 7d and inset). In the case of bisimide 1b, similar UV and fluorescence change was observed. Upon addition of TFA, the absorbance at 347, 408 and 432 nm was decreased while a new broad absorption at ca. 491 nm was appeared which indicated a cation was formed (Fig. 7a). The blue fluorescence at 443 nm and 464 nm was also quenched. However, a weak red orange emission around 550 nm was observed (Fig. 7b and inset). Note that there was no further spectral change even add more TFA and stand for overnight, indicating that the process is thermodynamic rather than kinetic.

|
Download:
|
| Fig. 7. UV–vis spectra change of TFA titration to PPIs 1b (a) and 2b (c). Fluoroesence spectra change of TFA titration to PPIs 1b (b) (λex = 330 nm) and 2b (d) (λex = 346 nm). The black arrow represents the change of absorbance and intensity as the amount of TFA increased. Inset: color change (a, c) and fluorescence quench (b, d) upon addition of TFA to the solution of 1b and 2b in CHCl3 (0.01 mg/mL). | |
{kind=link}
In summary, we report a new kind of n–channel semiconductors based on pyrene-phenazine fused bisimides and monoimide. The approach developed in the paper is facile and can be steadily extended to the utilization of other conjugated molecules as focal cores. The solution-process allows for an easy self-assembly to form high aspect ratio crystalline tapes. In the self-assembled state, the semiconductors show a high carrier mobility of 4.1 cm2 V-1 s-1 at 25 ℃. The N-terminals are ready for further chemical modification. For example, further tailoring hydrophobic, hydrophilic chains or integrating electron-donating units will allow for tuning self-assembly pattern, controlling redox potential, and thus realizing ambipolar charge carrier transport etc., which are now in progress in our laboratory.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 51522303, 21602154), National Key R & D Program of China (No. 2017YFA0207500), and the Thousand Youth Talents Plan.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.09.015.
| [1] |
(a) K. Müllen, U. Scherf, Organic Light Emitting-Devices: Synthesis, Properties and Applications, Wiley-VCH, Weinheim, 2006; (b) M. Pope, C. E. Swenberg, Electronic Processes in Organic Crystals and Polymers, Oxford University Press Inc., Oxford, 1999; (c) H. Inokuchi, Org. Electron. 7(2006) 62-76; (d) H. Klauk, Organic Electronics: Materials, Manufacturing and Applications, Wiley-VCH, Weinheim, 2006. |
| [2] |
(a) J. E. Anthony, Chem. Rev. 106(2006) 5028-5048; (b) A. R. Murphy, J. M. J. Fréchet, Chem. Rev. 107(2007) 1066-1096; (c) J. Zaumseil, H. Sirringhaus, Chem. Rev. 107(2007) 1296-1323. |
| [3] |
(a) Z. Bao, A. J. Lovinger, J. Brown, J. Am. Chem. Soc. 120(1998) 207-208; (b) Y. Sakamoto, T. Suzuki, A. Miura, et al., J. Am Chem. Soc. 122(2000) 1832-1833; (c) S. B. Heidenhain, Y. Sakamoto, T. Suzuki, et al., J. Am. Chem. Soc. 122(2000) 10240-10241; (d) Y. Sakamoto, K. Shingo, T. Suzuki, J. Am. Chem. Soc. 23(2001) 4643-4644; (e) A. Facchetti, M. H. Yoon, C. L. Stern, et al., J. Am. Chem. Soc. 126(2004) 13480-13501; (f) M. Fachetti, et al., Adv. Mater. 15(2003) 33-38. |
| [4] |
(a) M. Strukelj, F. Papadimitrakopoulos, T. M. Miller, et al., Science 267(1995) 1969-1972; (b) C. J. Tonzola, M. M. Alam, W. Kaminsky, et al., J. Am. Chem. Soc. 125(2003) 13548-13558; (c) T. W. Kwon, M. M. Alam, S. A. Jenekhe, Chem. Mater. 16(2004) 4657-4666. |
| [5] |
(a) B. C. Thompson, J. M. J. Fréchet, Angew. Chem. 120(2007) 62-82; (b) B. C. Thompson, J. M. J. Fréchet, Angew. Chem. Int. Ed. 47(2008) 58-77. |
| [6] |
(a) H. E. Katz, A. J. Lovinger, J. Johnson, et al., Nature 404(2000) 478-481; (b) L. L. Miller, K. R. Mann, Acc. Chem. Res. 29(1996) 417-423; (c) B. A. Gregg, R. A. Cormier, J. Am. Chem. Soc. 123(2001) 7959-7960; (d) B. A. Jones, A. Facchetti, M. R. Wasielewski, et al., J. Am. Chem. Soc. 129(2007) 15259-15278. |
| [7] |
(a) Z. Wang, C. Kim, A. Facchetti, et al., J. Am. Chem. Soc. 129(2007) 13362-13363; (b) X. K. Gao, Y. Wang, X. D. Yang, et al., Adv. Mater. 19(2007) 3037-3042; (c) F. Ilhan, D. S. Tyson, D. J. Stasko, et al., J. Am. Chem. Soc. 128(2006) 702-703. |
| [8] |
(a) B. Gao, M. Wang, Y. Cheng, et al., J. Am. Chem. Soc. 130(2008) 8297-8306; (b) D. C. Lee, K. Jang, K. K. McGrath, et al., Chem. Mater. 20(2008) 3688-3695. |
| [9] |
J. Hu, D. Zhang, S.Z.D. Cheng, et al., Chem. Mater. 16(2004) 4912-4915. DOI:10.1021/cm0492179 |
| [10] |
J. Cornil, V. Lemaur, J.P. Calbert, et al., Adv. Mater. 14(2002) 726-729. DOI:10.1002/1521-4095(20020517)14:10<726::AID-ADMA726>3.0.CO;2-D |
| [11] |
(a) L. Yu, M. Chen, L. R. Dalton, Chem. Mater. 2(1990) 649-659; (b) J. K. Stille, E. L. Mainen, Macromolecules 1(1968) 36-42; (c) T. Yamamoto, K. Sugiyama, T. Kushida, et al., J. Am. Chem. Soc. 118(1996) 3930-3937; (d) C. Y. Zhang, J. M. Tour, J. Am. Chem. Soc. 121(1999) 8783-8790. |
| [12] |
H. Du, R.A. Fuh, J. Li, et al., Photochem. Photobiol. 68(1998) 141-142. |
| [13] |
(a) K. Balakrishan, A. Datar, T. Naddo, et al., J. Am. Chem. Soc. 128(2006) 7390-7398; (b) Y. Che, D. Aniket, X. Yang, et al., J. Am. Chem. Soc. 129(2007) 6354-6355; (c) Y. Che, A. Datar, K. Balakrishan, et al., J. Am. Chem. Soc. 129(2007) 7234-7235. |
| [14] |
Y. Che, X. Yang, S. Loser, et al., Nano Lett. 8(2008) 2219-2223. DOI:10.1021/nl080761g |
| [15] |
(a) A. Acharya, S. Seki, Y. Koizumi, et al., J. Phys. Chem. B 109(2005) 20174-20179; (b) A. Saeki, S. Seki, T. Takenobu, et al., Adv. Mater. 20(2008) 920-923. |
| [16] |
(a) A. Acharya, S. Seki, Y. Koizumi, et al., J. Phys. Chem. B 109(2005) 20174-20179; (b) A. Saeki, S. Seki, T. Takenobu, et al., Adv. Mater. 20(2008) 920-923; (c) Y. Yamamoto, T. Fukushima, Y. Suna, et al., Science 314(2006) 1761-1764; (d) T. Amaya, S. Seki, T. Moriuchi, et al., J. Am. Chem. Soc. 131(2009) 408-409; (e) W. S. Li, Y. Yamamoto, T. Fukushima, et al., J. Am. Chem. Soc. 130(2008) 8886-8887. |
| [17] |
D.C. Lee, K.K. McGrath, K. Jang, Chem. Commun.(2008), 3636-3638. |
| [18] |
J. Hu, D. Zhang, F.W. Harris, J. Org. Chem. 70(2005) 707-708. |