 2018, Vol. 29
2018, Vol. 29 



The exhausting petroleum resources and serious air pollution caused by burning of fossil fuel promote people to look for new kinds of clean energy [1], among which biodiesel has arisen increasing attention due to its advantages such as renewable, degradable and pollution-free [2-4]. Preparation methods of biodiesel can be divided into physical, chemical and biological method. The main defect of physical method is that its product quality cannot achieve the user's requirement and the biological method has disadvantages such as poor stability and long production cycle. The main chemical methods include splitting method and transesterification method [5]. Most attention has been focused on the transesterification method because coke and carbon deposition problem is difficult to be dissolved in splitting method [6].
Transesterification reaction is reversible, thus, to achieve complete transesterification reaction, low boiling point alcohol is generally replaced with high boiling point alcohol and the low boiling point alcohol must be separated timely. However, when biodiesol is prepared through transesterification reaction, methanol or ethanol is used to replace glycerol in vegetable oils, which is unfavorable to complete the transesterification reaction. The main chemical composition of vegetable oils is triglyceride of fatty acids. Incompolete transesterification reaction would result in the generation of mono-and di-glyceride of fatty acids, both of which have surface activity and lead to higher glycerol content in the products [7].
As it is well known, the technology of obtaining glycerol and fatty acids from oil hydrolysis has been well developed and industrialized. Consequently, based on the oil hydrolysis technology, an ideal preparation method of biodiesol is direct esterification of fatty acid with low-carbon alcohol. The existing problems in transesterification method can be overcome in direct esterification method [8, 9]. A catalyst is needed in the esterification reaction of fatty acid and alcohol. Solid acid catalysts receive more and more attention recent years because they have many advantages including easy separation, pollution-free and no corrosion [2, 10-14].
Tungstophosphoric acid (H3PW12O40·xH2O, HPW) has been supported onto SiO2, TiO2, ZrO2, Nb2O5, Al2O3, activated carbon, and MCM-41 molecular sieve to obtain solid acid catalysts [15-18]. ZrO2 is a kind of catalyst carrier with excellent properties [19] and the HPW/ZrO2 catalysts have been widely used in many kinds of reaction including esterification and transesterification [20-22], isomerization [23], oxidation [24], bromization [25], alkylation [26], glycerol dehydration [27] and so forth.
Structure and morphology of the carrier have significant influence on the catalytic activity. Zirconia nanotube arrays, prepared by anodization method, have large specific surface area and their tubular structure can act as nano reactors. Therefore, when used as carrier, ZrO2 nanotube arrays might endow the catalyst better performance [28, 29]. However, up to now, few studies have been done on the application of ZrO2 nanotube arrays as HPW catalyst carrier. In this paper, HPW was supported onto ZrO2 nanotube arrays to form zirconia nanotube-supported HPW catalyst for the first time. The influences of various factors on the catalytic activity have been discussed in detail. The catalytic activity on the esterification reaction between ethanol and the main fatty acids in vegetable oil (oleic acid, stearic acid and lauric acid) has also been studied.
In a typical experiment, zirconia nanotube arrays were prepared by anodization method in the mixture of formamide and glycerol (volume ratio = 1:1) containing 1 wt% NH4F and 1 wt% H2O. Zirconium foils were used as anodic electrode while platinum foil was used as cathodic electrode. The distance between anodic and cathodic electrodes was 20 mm. All anodization experiments were carried out at room temperature. Anodization was conducted for 3 h at 50 V. During the experiments, the solutions were stirred using a magnetic stirrer. After the anodization, nanotube arrays were peeled off from zirconium foil, rinsed with water and dried at 100 ℃ to obtain the zirconia nanotube carrier. The carrier was annealed for 4 h at a certain temperature, followed by ultrasonic impregnation for 60 min in ethanol solution of HPW, and then dried and calcined for 4 h to prepare zirconia nanotube-supported HPW catalyst.
The morphologies of the nanotube and catalyst samples were observed through Philips XL 30 TMP scanning electron microscope (SEM, 20 kV accelerating voltage). Crystal phase analysis of the samples was conducted through Philips X' pert MPD X-ray powder diffraction analyzer (XRD, copper target, 50 kV, 40 mA, ). The samples were also characterized by Fourier transform infrared spectrometer (FTIR, WQF-410, China) and thermo analyzer (SDT 2960, TA Instruments, USA).
The catalytic activity experiments were carried out in a threenecked flask equipped with thermometer, stirrer and reflux condenser. In a typical reaction, the reactant consisted of 0.20 mol absolute ethyl alcohol, 0.10 mol RCOOH (propionic acid, lauric acid and oleic acid) and 0.33 g catalyst. The reaction solution was refluxed at 90 ℃ for 120 min. After the reaction, the product was cooled and the RCOOH conversion percentage was determined.
Zirconia nanotube arrays were annealed at 400 ℃ and then loaded with different amount of HPW, followed by calcining at 200 ℃ to prepare the catalysts. Fig. 1 shows the morphologies of zirconia nanotube arrays (Fig. 1a) and the catalysts with different loading amount (Figs. 1b-d). As shown in Fig. 1, when the loading amount of HPW is 20% and 35% (weight percentage, similarly hereinafter), the surface of nanotube arrays is covered with some scattered particles. Accumulated particles are observed on the catalyst surface when the loading amount of HPW is 55%.

|
Download:
|
| Fig. 1. SEM images of zirconia nanotube arrays (a) and the catalyst loaded with 20% HPW (b); 35% HPW (c); 55% HPW (d). | |
{kind=link}
Fig. 2A shows the X-ray diffraction patterns of the nanotube arrays (Fig. 2A, curve a), catalysts (Fig. 2A, curves b-d) and HPW (Fig. 2A, curve e). The XRD patterns reveal that zirconia nanotube arrays are of two mixed crystal structure (monoclinic and tetragonal phase) after annealing (Fig. 2A, curve a). The main diffraction peaks of HPW (Fig. 2A, curve e) at 2θ = 10.4°, 20.8°, 23.2°, 25.5°, 29.5°, 37.8° still exist in the catalysts (Fig. 2A, curves b-d) and the intensities of these peaks increase along with the HPW loading amount, indicating that the main structure (Keggin structure) of HPW in the catalysts has no obvious change. However, compared with pure HPW, the diffraction peaks of HPW at 2θ = 16.2°, 18.6°, 19.5° are hardly found and the diffraction peak at 2θ = 10.4° splits and becomes wider evidently in the catalysts (Fig. 2A, curves b-d). The phenomena indicate that there are strong interactions between HPW and zirconia nanotube arrays, which change the microstructure of HPW and decrease its long-range order and reduce the crystal size. FTIR spectra of the nanotubes (Fig. 2B, curve a), catalysts (Fig. 2B, curves b-d) and HPW (Fig. 2B, curve e) are shown in Fig. 2B. HPW displays strong absorption peaks at 1080 cm-1 and 985 cm-1, corresponding to P—O and W—O vibration adsorption in Keggin structure (Fig. 2B, curve e), while zironia nanotubes have no adsorption peak at these two positions (Fig. 2B, curve a). In the catalysts, characteristic absorption peaks (1080 cm-1 and 985 cm-1) of HPW almost keep unchanged, while the intensities of absorption peaks increase with the loading amount (Fig. 2B, curves b-d), indicating that the main structure of HPW has no obvious changes, which is in agreement with the XRD results.

|
Download:
|
| Fig. 2. XRD patterns (A) and Infrared spectra (B) of the samples. a: ZrO2 nanotube arrays; b: 20% HPW/ZrO2; c: 35% HPW/ZrO2; d: 55% HPW/ZrO2; e: HPW. | |
{kind=link}
The catalytic activity experiments indicate that HPW loading amount affects the conversion percentage markedly. With the increasing loading amount, the conversion percentage increases and when the loading amount is over 35%, it has no obvious effect on the conversion percentage (Fig. S1 in Supporting information). With relatively lower loading amount, the contact area between nanotubes and HPW increases with increasing loading amount and coverage extent, more active sites form and the catalytic activities elevate. When the loading amount achieves saturation, more loading amount would not increase the contact area and active sites. Therefore, the conversion percentage almost remains unchanged when the loading amount is over 35% [21].
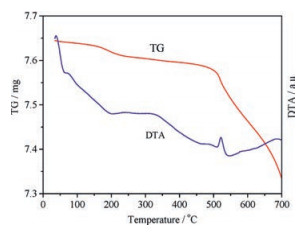
The carrier pretreatment temperature also has apparent effects on catalytic activity. The catalytic activity first increases and then decreases slightly along with the increasing temperature, reaching the highest catalytic activity at 400 ℃ (Fig. S2 in Supporting information). Fig. 3 gives the thermogravimetric (TG) and differential thermal analysis (DTA) curves of the zirconia nanotube carrier. As shown in the TG curve, the sample has remarkable weight loss below 200 ℃ or above 500 ℃, and its weight remains almost constant between 200 ℃ and 500 ℃. The DTA curve shows that an exothermic process appears between 200 ℃ and 400 ℃ and there is an exothermic peak at 520 ℃. The weight loss below 200 ℃ corresponds to the elimination of adsorbed water. The structure order degree of zirconia increases with the rising temperature and zirconia starts to crystallize at 400 ℃. The condensation dehydration of hydroxyl groups at the zirconia surface takes place when the temperature is higher than 500 ℃, and crystal phase transformation appears at 520 ℃.

|
Download:
|
| Fig. 3. TG-DTA spectra of zirconia nanotube carrier. | |
{kind=link}
The TG-DTA results show that along with the increasing temperature, the adsorbed water is eliminated and the structure order degree rises, which favors the combination of HPW with zirconia carrier and is conducive to the increasing catalytic activity. When the temperature is too high, the catalytic activity decreases because of worse combination between HPW and carrier, which can be ascribed to some unfavorable factors including the condensation dehydration of surface hydroxyl groups, crystal phase transformation, larger grain size and lower specific surface area [30].
The influence of catalyst calcination temperatures on the catalytic activity is similar with that of carrier pretreatment temperature. The conversion percentage first rises slightly, reaching the highest catalyst activity at 200 ℃, and then drops with rising calcination temperature (Fig. S3 in Supporting information). When the catalysts are calcined at 200 ℃, the rising catalytic activities might be ascribed to the water loss. After that, further increasing the calcination temperatures would lead to the decreasing catalytic activities due to the decomposition of HPW [21].
According to the results discussed above, zirconia nanotube arrays were pretreated at 400 ℃ and loaded with 35% HPW, followed by calcinating at 200 ℃ to obtain the catalyst. Catalytic syntheses of ethyl propionate, ethyl laurate and ethyl oleate were conducted using the as-prepared catalyst. The influence of catalyst dosage on RCOOH conversion percentage is depicted in Fig. 4. The conversion percentage increases with the rising catalyst dosage and becomes stable when the catalyst dosage reaches 5% (the conversion percentage of propionic acid, lauric acid and oleic acid is 66.6%, 78.1% and 86.3%, respectively).

|
Download:
|
| Fig. 4. Influence of catalyst dosage. | |
{kind=link}
As shown in Fig. 4, the conversion percentage increases with the increasing alkyl chain length, indicating that the catalyst is more suitable to synthesize higher fatty acid ester. Under the optimal conditions (reaction temperature:120 ℃; reaction time: 3 h; fatty acid/ethanol molar ratio: 1/12), the conversion percentages of lauric acid, oleic acid and stearic acid are all higher than 98.5%. In addition, the conversion percentage is still above 90% after recycling 3 times. Compared with the similar catalysts prepared through loading HPW onto zirconia nanoparticles [16, 21, 31], the catalysts prepared in this work exhibit better catalytic activity. This might be ascribed to the following reasons: firstly, the hollow nanotubes would provide transfer channel for the reactants and increase effective specific surface area of the catalysts; secondly, there are much stronger interactions between HPW and ZrO2 because of the lattice disorder and more defects in ZrO2 resulting from the nanotube structure.
In summary, zirconia nanotube-supported HPW catalysts were prepared through anodization and ultrasonic impregnation method. The pretreated temperature of the zirconia nanotubes, HPW loading amount, and calcination temperature of the catalyst have significant influences on the catalytic activity. The catalyst prepared through pretreating zirconia nanotubes at 400 ℃, loading with 35% HPW, and calcinating at 200 ℃ is highly active for the esterification reaction between alcohol and fatty acid. Under the reaction temperature of 120 ℃, reaction time of 3 h, and fatty acid/ ethanol molar ratio of 1/12, the conversion percentages of lauric acid, oleic acid and stearic acid are all higher than 98.5%.
AcknowledgmentsThis work is supported by the National Natural Science Foundation of China (No. 51272064) and Key Basic Research Program of Hebei Province of China (No. 17964401D).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.08.047.
| [1] |
J.X. Wang, K.T. Chen, S.T. Huang, C.C. Chen, Chin. Chem. Lett. 22(2011) 1363-1366. |
| [2] |
K. Jacobson, R. Gopinath, L.C. Meher, A.K. Dalai, Appl. Catal. B-Environ. 85(2008) 86-91. |
| [3] |
P. Campanelli, M. Banchero, L. Manna, Fuel 89(2010) 3675-3682. DOI:10.1016/j.fuel.2010.07.033 |
| [4] |
H. Mootabadi, B. Salamatinia, S. Bhatia, A.Z. Abdullah, Fuel 89(2010) 1818-1825. DOI:10.1016/j.fuel.2009.12.023 |
| [5] |
S. Zhou, L. Liu, B. Wang, F. Xu, R.C. Sun, Chin. Chem. Lett. 23(2012) 379-382. DOI:10.1016/j.cclet.2012.01.034 |
| [6] |
F. Ma, M.A. Hanna, Bioresour. Technol. 70(1999) 1-15. DOI:10.1016/S0960-8524(99)00025-5 |
| [7] |
Y. Sharma, B. Singh, S. Upadhyay, Fuel 87(2008) 2355-2373. DOI:10.1016/j.fuel.2008.01.014 |
| [8] |
M. Veillette, A. Giroir-Fendler, N. Faucheux, M. Heitz, J. Chem. Eng. 308(2017) 101-109. DOI:10.1016/j.cej.2016.07.061 |
| [9] |
A. Yousuf, M.R. Khan, M.A. Islam, Z. Ab Wahid, D. Pirozzi, Biotechnol. Lett. 39(2017) 13-23. DOI:10.1007/s10529-016-2217-x |
| [10] |
N. Laosiripojana, W. Kiatkittipong, W. Sutthisripok, S. Assabumrungrat, Bioresour. Technol. 101(2010) 8416-8423. |
| [11] |
D.E. Lopez, J.G. Goodwin, D.A. Bruce, S. Furuta, Appl. Catal. A-Gen. 339(2008) 76-83. DOI:10.1016/j.apcata.2008.01.009 |
| [12] |
Y.M. Park, S.H. Chung, H.J. Eom, J.S. Lee, K.Y. Lee, Bioresour. Technol. 101(2010) 6589-6593. DOI:10.1016/j.biortech.2010.03.109 |
| [13] |
S.B. Yu, H.J. Zang, X.L. Yang, et al., Chin. Chem. Lett. 28(2017) 1479-1484. DOI:10.1016/j.cclet.2017.02.016 |
| [14] |
M. Zabeti, W.M.A. Wan Daud, M.K. Aroua, Fuel Process. Technol. 90(2009) 770-777. |
| [15] |
J.W. Ding, R. Wang, Chin. Chem. Lett. 27(2016) 655-658. DOI:10.1016/j.cclet.2016.03.005 |
| [16] |
E. Sert, F.S. Atalay, Ind. Eng. Chem. Res. 51(2012) 6666-6671. DOI:10.1021/ie202609f |
| [17] |
L. Pizzio, C. Caceres, M. Blanco, Appl. Catal. A-Gen. 167(1998) 283-294. DOI:10.1016/S0926-860X(97)00328-1 |
| [18] |
M.J. Verhoef, P.J. Kooyman, J.A. Peters, H. van Bekkum, Micropor. Mesopor. Mat. 27(1999) 365-371. DOI:10.1016/S1387-1811(98)00269-8 |
| [19] |
Y.F. Ma, H. Wang, G.Y. Xu, Chin. Chem. Lett. 28(2017) 1153-1158. |
| [20] |
M.G. Kulkarni, R. Gopinath, L.C. Meher, A.K. Dalai, Green Chem. 8(2006) 1056-1062. |
| [21] |
C.F. Oliveira, L.M. Dezaneti, F.A.C. Garcia, et al., Appl. Catal. A-Gen. 372(2010) 153-161. |
| [22] |
G. Sunita, B.M. Devassy, A. Vinu, et al., Catal. Commun. 9(2008) 696-702. DOI:10.1016/j.catcom.2007.08.007 |
| [23] |
A. Alsalme, E.F. Kozhevnikova, I.V. Kozhevnikov, Appl. Catal. A-Gen. 390(2010) 219-224. DOI:10.1016/j.apcata.2010.10.018 |
| [24] |
K. Parida, S. Mallick, Catal. Commun. 11(2009) 51-57. DOI:10.1016/j.catcom.2009.07.002 |
| [25] |
S. Mallick, K. Parida, Catal. Commun. 8(2007) 889-893. DOI:10.1016/j.catcom.2006.09.016 |
| [26] |
B.M. Devassy, G. Shanbhag, F. Lefebvre, et al., J. Mol. Catal. A-Chem. 230(2005) 113-119. DOI:10.1016/j.molcata.2004.12.028 |
| [27] |
S.H. Chai, H.P. Wang, Y. Liang, B.Q. Xu, Appl. Catal. A-Gen. 353(2009) 213-222. DOI:10.1016/j.apcata.2008.10.040 |
| [28] |
X.M. Liu, G. Lu, Z.F. Yan, Appl. Catal. A-Gen. 279(2005) 241-245. |
| [29] |
N. Lucas, A. Bordoloi, A.P. Amrute, et al., Appl. Catal. A-Gen. 352(2009) 74-80. DOI:10.1016/j.apcata.2008.09.032 |
| [30] |
E. Lopez-Salinas, J. Hernandez-Cortez, I. Schifter, et al., Appl. Catal. A-Gen. 193(2000) 215-225. DOI:10.1016/S0926-860X(99)00431-7 |
| [31] |
M.G. Kulkarni, R. Gopinath, L.C. Meher, A.K. Dalai, Green Chem. 8(2006) 1056-1062. DOI:10.1039/b605713f |