 2018, Vol. 29
2018, Vol. 29 



b Hunan Province Cooperative Innovation Center for Molecular Target New Drug Study, University of South China, Hengyang 421001, China;
c Key Laboratory of Tropical Marine Bio-resources and Ecology, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou 510301, China;
d Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 610054, China
Biofilms are complex microbial communities adhere to moist surfaces. Chemically it is a mixture of self-produced polysaccharide, proteins and DNA [1]. Biofilms are frequently associated with chronic refractory infections, which are extremely resistant to antibiotics, disinfectants, as well as human immune system [2]. Due to the adhesion of biofilm-forming microorganism to surfaces, biofilm-associated infections (BAI) often occur in patients who receive medical devices and implants [3]. This increases the incidence of nosocomial infections and poses a clinical challenge.
So far, there is still paucity of effective treatment options for BAI because biofilms are extremely difficult to be eradicated [4]. As the population of susceptible patients with indwelling medical devices is mounting up [5], there is a critical need to develop new antimicrobial compounds for preventing BAI. Biofilm and planktonic growth are two distinctive and reversible bacterial growth phases. Biofilm-grown cells exhibit increased tolerance to environmental stresses [6]. Although there are various types of potential anti-biofilm agents (antimicrobial peptides, nanoparticles, berberine, furanone derivatives, etc.), none of these has been used clinically so far. Antimicrobials kill bacteria at bactericidal concentrations. However, once the concentration is lower than the minimal bactericidal concentration, the survived bacteria can gradually adapt to the hostile environment and form biofilm [7, 8]. Therefore, an ideal anti-biofilm agent should inhibit bacterial adhesion and forming biofilm rather than just inhibiting or killing the bacteria [9, 10, 11].
Within the biofilm environment, inter-bacterial communication is based on self-generated signal molecule known as autoinducers (AIs). AIs accumulate in the microenvironment allow the bacteria to sense their cell numbers and alter gene transcription in order to coordinates expression of biofilm-forming property, virulence, and antibiotic resistance [12, 13]. Currently, AIs and its corresponding receptors have been recognized as important therapeutic targets for biofilm-associated and drugresistant infections [14]. However, it remains a challenge to develop novel molecules that can block the signaling receptors of bacteria or inhibit the biosynthesis of AIs [15]. QS signaling molecules, such as acyl-homoserine lactones (AHLs) and its analogues, can inhibit biofilm but have no effect on viability of bacteria. These agents are considered as potential antibiofilm agents. Furanone derivatives, such as halogenated furanone and patulin, have been shown to possess QS antagonizing activity and biofilm inhibitory effects [16]. Previous study also showed that furanone derivatives interfered with QS signaling of Pseudomonas aeruginosa by binding to the QS receptor, such as LasR [17]. So far, there is no report on QS quenching potential of phenyl-substituted furanone derivatives. This study aimed to evaluate the antibiofilm activities of phenyl-substituted furanone derivatives, and to assess the interference of it with bacterial QS. Here, the antibiofilm and QS-quenching activities of a furanone derivative, homogentisic acid γ-lactone, are described (Scheme S1 in Supporting information).
Chemicals used in this study were as follows. Homogentisic acid γ-lactone (5-hydroxy-2(3H)-benzofuranone, HgAL), N-butyryl-DL-homoserine lactone (C4-HSL, C8H13NO3, CAS No. 98426-48-3), N-(3-oxotetradecanoyl)-L-homoserine lactone (3-oxo-C12-HSL, C16H27NO4, CAS No. 168982-69-2) were purchased from SigmaAldrich (China). Mueller-Hinton broth (MHB) and Mueller-Hinton agar (MHA) were purchased from Oxoid (Basingstoke, UK). Doubledeionized water was used for preparing all aqueous solutions. All chemicals used in the experiments were of analytical grade and were used without further purifications.
HgAL used in this study is a benzofuranone derivatives and with low solubility in water (< 350 μg/mL) at room temperature. To prepare HgAL stock solution, HgAL powder was dissolved in a small volume of DMSO and filtered through a 0.22-μm pore membrane filter. DMSO concentrations were maintained below 0.5% (v/v) to avoid physiological toxicity [18, 19]. The HgAL stock solution was then diluted in MHB to concentrations ranged from 0 to 400 μg/mL.
Pseudomonas aeruginosa PAO1 strain was used in this study. A single colony of P. aeruginosa grown on MHA plate was inoculated into 10 mL fresh MHB and incubated overnight at 37 ℃ with shaking at 200 rpm. The overnight bacterial culture was inoculated into the MHB-diluted HgAL solutions (at various concentrations) to a final density of 1.0 × 105 colony-forming unit (CFU)/mL [20]. Bacterial suspensions prepared ("initial suspensions") were used for subsequent experiments.
Confocal laser scanning microscopy (CLSM) was used to evaluate the anti-biofilm effect of HgAL. Detailed steps were as follows. Five milliliters of P. aeruginosa initial suspensions were dispensed into each well of a 6-well plastic plate (with an 8-mm diameter glass coverslips placed at the bottom). After incubating at 37 ℃ for 48 h, the coverslips were gently washed with phosphatebuffered saline (PBS, pH 7.4) three times and fixed with 4% paraformaldehyde for 4 h at 4 ℃. After rinsing twice in PBS and airdrying, the coverslips were stained with 200 μL of 1 mg/mL acridine orange for 20 min at 4 ℃ in the dark. Stained coverslips were gently washed twice with PBS, air-dried and examined using CLSM.
Besides, biofilm formation on silicon slide was examined by scanning electron microscopy (SEM). Briefly, 2 mL of the P. aeruginosa initial suspensions were dispensed into each well of a 12-well plastic plate (pre-loaded with a 1 cm × 1 cm silicon slide). After incubating at 37 ℃ for 48 h, the silicon slides were gently washed three times with PBS, fixed with anhydrous methanol for 3 min, dried at 37 ℃, dehydrated in increasing concentrations of ethanol solutions (30%, 50%, 70%, 80%, 90%, 95% and 100%), lyophilized in a vacuum freeze-drier, and examined under SEM (TESCAN VEGA3).
To figure out whether HgAL can inhibit the biosynthesis of QS signal molecules in P. aeruginosa, high performance liquid chromatography (HPLC) analysis of C4-HSL and 3-oxo-C12-HSL was carried out. One hundred millimeters of P. aeruginosa initial suspension were dispensed to conical flasks (250 mL) and incubated at 37 ℃ for 72 h under static conditions. After centrifuging at 5000 × g for 15 min, the supernatant was extracted twice in 50 mL ethyl acetate. The extract was dried at 50 ℃ in rotary evaporators. The residue was re-suspended in 2 mL methanol in an ice bath. After filtering through the 0.22 μm hydrophilic millipore filter, the concentration of C4-HSL and 3-oxo-C12-HSL was measured using HPLC according to a previous study [21, 22] with appropriate modifications. The standards (C4-HSL and 3-oxo-C12-HSL) were prepared by methanol to concentrations of 75 μg/mL and 60 μg/mL. The regular-phase silica gel C18 column (150 mm × 4.6 mm i.d., S-5 μm) was used with acetonitrile-water (with 0.1% formic acid) at a flow rate of 1.0 mL/min. The injection volume was 20 μL and 210 nm was the detection wavelength. The acetonitrile gradient was set as follows: 0 to 35 min, 5%–100%, 35 to 45 min, 100%; 45 to 50 min, 100%–5%; 50 to 60 min, 5%.
Results of bacterial toxicity assays of HgAL against P. aeruginosa are shown in Fig. S1 (Supporting information). The results of this study showed that HgAL had no antimicrobial activity against P. aeruginosa at a concentration lower than 160 mg/mL. This result suggests that the biological effects of HgAL mentioned below were not be caused by the decline in bacterial counts.
Bacterial extracellular matrix (ECM) is an important biofilm structural component as it plays roles in gluing and shielding bacterial cells in the biofilm [23-25]. Due to the protection of ECM, biofilm-associated bacteria are difficult to be eradicated by chemotherapeutic agents [26]. Hence, we believed that disassembling ECM facilitates the elimination of biofilm-protected bacteria and decrease the incidence of BAI. Extracellular matrix protein (EMP) and extracellular polysaccharides are two important components of ECM. Pathogens like Staphylococcus aureus and P. aeruginosa usually secrete high levels of EMP which contribute to bacterial adhesion and biofilm formation [27]. It is shown in Fig. S2A (Supporting information) that the EMP concentrations have decreased after treating with different concentrations of HgAL comparing to that of the untreated group. At a treatment concentration of 160 μg/mL, the amounts of EMP secreted were inhibited remarkably. With similar encouraging results, HgAL treatment led to a significant decrease in the total polysaccharides concentrations (Fig. S2B in Supporting information). It is evident that HgAL effectively reduces the synthesis of ECM in a dosedependent manner. Actually, the bacterial EMP is closely related to the bacterial adherence, which can be indirectly evaluated by determining the cell-surface hydrophobicity. Microbial adhesion to hydrocarbons (MATH) assay [28, 29] is a common way to detect the cell-surface hydrophobicity. In Fig. S3A (Supporting information), the percentage of hydrophobicity of PAO1 strain was dramatically reduced after treating with different concentrations of HgAL, suggesting that HgAL can down-regulate the adhesion abilities of P. aeruginosa.
Moreover, pyocyanin is one of the many toxins secreted by P. aeruginosa. Toxicity of pyocyanin is due to its ability to oxidize and reduce other molecules, making it toxic to other microbes and the airway epithelium infected by P. aeruginosa during cystic fibrosis [30, 31]. In Fig. S3B (Supporting information), production of pyocyanin decreased significantly after treatment by HgAL at concentrations above 50 μg/mL.
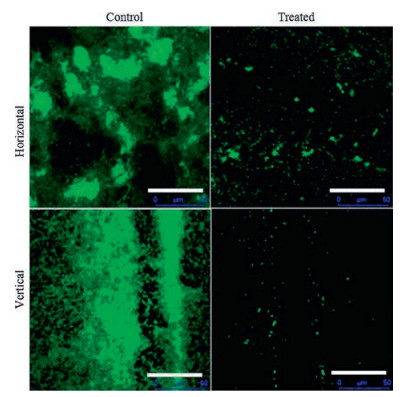
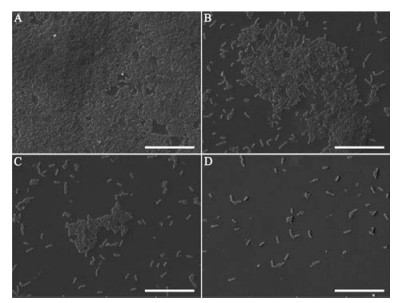
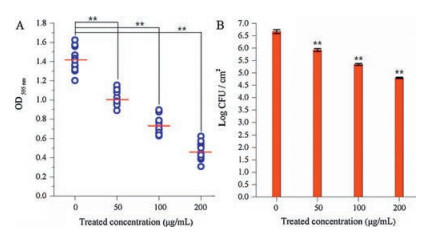
To investigate the anti-biofilm activity of HgAL against P. aeruginosa, biofilm morphology was examined under CLSM. The amount of biofilm biomass was quantified using crystal violet (CV) assay, viable bacteria in the biofilm was also counted. Fig. 1 shows the distributions of biofilm formed on cover slips surfaces by the HgAL-treated and untreated P. aeruginosa. After growth for 48 h, the HgAL-treated P. aeruginosa biofilm exhibited smaller aggregates of microorganisms and looser structure compared to the untreated P. aeruginosa. These results suggest that biofilm formation on the upper surface of liquid or at the bottom section can be inhibited by treatment of 100 μg/mL HgAL. When examined under SEM, untreated P. aeruginosa formed a sufficiently thick layer of bacterial cells after incubation for 48 h (Fig. 2A). At 50 μg/mL of HgAL, the levels of cell aggregation greatly reduced and the biofilms were much thinner than the untreated biofilm (Fig. 2B). At 100 μg/mL (Fig. 2C) and 150 μg/mL (Fig. 2D) of HgAL, only a few small microcolonies and single bacterial cells were seen, indicating that no or small amount of biofilm was formed. Thus, the SEM results further demonstrated that biofilm-forming ability of P. aeruginosa was reduced in the presence of HgAL. The crystal violet (CV) assay results showed that biofilm-forming ability of P. aeruginosa was significantly reduced by HgAL treatment in a dose-dependent manner (Fig. 3A). To obtain more accurate experimental evidence, the numbers of viable bacteria present in the biofilms were counted. As shown in Fig. 3B, the amounts of biofilm bacteria were significantly reduced after treating with different concentrations of HgAL. All results of biofilm detection illustrated that HgAL has anti-biofilm activity against P. aeruginosa in a dose-dependent manner.

|
Download:
|
| Fig. 1. CLSM analysis of P. aeruginosa biofilms formed by untreated (left) and treated (right) with 100 μg/mL HgAL during 48 h growth. Biofilms were stained with acridine orange, resulting in all bacteria appearing green (including live and dead bacteria). Scale bars are 50 μm. | |
{kind=link}

|
Download:
|
| Fig. 2. SEM images of biofilms formed by P. aeruginosa incubated with 0 (A); 50 (B); 100 (C); and 150 (D) μg/mL HgAL for 48h. Scale bars: 20 μm. | |
{kind=link}

|
Download:
|
| Fig. 3. Absorbance at 595 nm of CV-stained P. aeruginosa biofilms treated with different concentrations of HgAL (A). Viable bacterial counts of P. aeruginosa biofilms treated with different concentrations of HgAL (B). | |
{kind=link}
Quorum sensing of several extracellular virulence factors and biofilm formation is controlled by N-acyl-homoserine lactone (acyl-HSL) signals in P. aeruginosa [32]. In this particular bacterial species, biofilm formation was mainly controlled by cell-to-cell signaling based on the secretion of 3-oxo-C12-HSL and C4-HSL [33]. In order to confirm the anti-QS activity of HgAL, both AHLs from P. aeruginosa biofilms were extracted directly and measured in the absence or presence of HgAL. The results confirmed that HgAL inhibited production of 3-oxo-C12-HSL (Fig. 4A) and C4-HSL (Fig. 4B). These results explained the mechanism of biofilm inhibition of HgAL observed in our study. As described above, HgAL effectively decreased the expression of bacterial EMP and exopolysaccharides, reduced bacterial adhesive ability, suppressed production of pyocyanin, and inhibited biofilm formation. All these changes were probably due to the interference effects on the QS signaling circuit. The basic QS circuit machinery consists of three elements, namely the signal synthase, signal receptor, and signal molecules. Through complex global regulatory networks, bacteria are able to respond to the surroundings through production of diffusible signal molecules. Thus, it is possible that HgAL inhibits expression of the QS-dependent target genes by quenching its QS signal molecules. In P. aeruginosa, LasI and RhlI synthesize the AIs 3-oxo-C12-HSL [34] and C4-HSL [35], respectively. The action mechanism of HgAL was proposed in Scheme S1. However, whether HgAL can inhibit the expression of QS synthases and synthase-related genes warrants further investigation.

|
Download:
|
| Fig. 4. The biosynthesis of QS AI molecules (A: 3-oxo-C12-HSL; B: C4-HSL) from P. aeruginosa treated with different concentrations of HgAL. | |
{kind=link}
In summary, this study showed that HgAL suppressed the expression of QS-dependent virulence factors of P. aeruginosa by quenching its QS signal molecules, making it an effective biofilm inhibitor and a potential alternative for treatment of biofilmassociated infections.
AcknowledgmentsThis study was supported by the National Natural Science Foundation of China (Nos. 81401512, 41606186), the Scientific Research Fund of Hunan Provincial Education Department (No. 14B157), China Postdoctoral Science Foundation (No. 2016M602419), and the Hong Kong Scholars Program (No. XJ2015022).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2017.09.052.
| [1] |
N. Høiby, T. Bjarnsholt, M. Givskov, et al., Int. J. Antimicrob. Agents 35(2010) 322-332. DOI:10.1016/j.ijantimicag.2009.12.011 |
| [2] |
T. Abee, T. Á.Kovács, O.P. Kuipers, S. van der Veen, Curr. Opin. Biotechnol. 22(2011) 172-179. DOI:10.1016/j.copbio.2010.10.016 |
| [3] |
N. Høiby, T. Bjarnsholt, C. Moser, et al., Clin. Microbiol. Infect. 21(2015) S1-S25. |
| [4] |
S. Silva, C.F. Rodrigues, D. Araújo, M.E. Rodrigues, M. Henriques, J. Fungi 3(2017) 8-24. DOI:10.3390/jof3010008 |
| [5] |
A.S. Lynch, D. Abbanat, Expert. Opin. Ther. Pat. 20(2010) 1373-1387. |
| [6] |
K. Lewis, Antimicrob. Agents Chemother. 45(2001) 999-1007. DOI:10.1128/AAC.45.4.999-1007.2001 |
| [7] |
H. Wu, C. Moser, H. Wang, N. Høiby, Z. Song, Int. J. Oral Sci. 7(2015) 1-7. |
| [8] |
D.L. Gomes, R.S. Peixoto, E.A. Barbosa, et al., J. Med. Microbiol. 62(2013) 754-760. DOI:10.1099/jmm.0.052373-0 |
| [9] |
O. Rendueles, J.B. Kaplan, J.M. Ghigo, Environ. Microbiol. 15(2013) 334-346. |
| [10] |
M. Kostakioti, M. Hadjifrangiskou, S.J. Hultgren, Cold Spring Harb. Perspect. Med. 3(2013) a010306. |
| [11] |
T. Zhang, C. Li, Y. Tian, et al., Chin. Chem. Lett. 28(2017) 1737-1742. DOI:10.1016/j.cclet.2017.05.022 |
| [12] |
S.T. Rutherford, B.L. Bassler, Cold Spring Harb. Perspect. Med. 2(2012) a012427. |
| [13] |
A. Deep, U. Chaudhary, V. Gupta, J. Lab Phys. 3(2011) 4-11. |
| [14] |
R.S. Smith, B.H. Iglewski, J. Clin. Invest. 112(2003) 1460-1465. DOI:10.1172/JCI200320364 |
| [15] |
B. LaSarrea, M.J. Federle, Microbiol. Mol. Biol. Rev. 77(2013) 73-111. DOI:10.1128/MMBR.00046-12 |
| [16] |
E.Y. Trizna, E.N. Khakimullina, L.Z. Latypova, et al., Acta Nat. 7(2015) 102-107. |
| [17] |
C. Kim, J. Kim, H.Y. Park, et al., Appl. Microbiol. Biotechnol. 80(2008) 37-47. |
| [18] |
T.A. Ben, N. Kitabatake, E. Doi, Agric. Biol. Chem. 54(1990) 2961-2966. |
| [19] |
S. Li, Z. Wang, Y. Wei, et al., Biomaterials 34(2013) 902-911. |
| [20] |
C.E. Stager, J.R. Davis, M.N. Saccomani, et al., Curr. Microbiol. 17(1988) 243-247. |
| [21] |
T. Kawaguchi, Y.P. Chen, R.S. Norman, A.W. Decho, Appl. Environ. Microbiol. 74(2008) 3667-3671. DOI:10.1128/AEM.02869-07 |
| [22] |
R. Tandel, N. Teradal, A. Satpati, S. Jaldappagari, Chin. Chem. Lett. 28(2017) 1429-1437. DOI:10.1016/j.cclet.2016.11.028 |
| [23] |
A. Dragoš, T. Á.Kovács, Trends Microbiol. 25(2017) 257-266. |
| [24] |
D. Davies, Nat. Rev. Drug Discov. 2(2003) 114-122. DOI:10.1038/nrd1008 |
| [25] |
N.K. Archer, M.J. Mazaitis, J.W. Costerton, et al., Virulence 2(2011) 445-459. |
| [26] |
J.L. Del Pozo, R. Patel, N. Engl. J. Med. 361(2009) 787-794. DOI:10.1056/NEJMcp0905029 |
| [27] |
S. Li, C. Wu, X. Tang, et al., Sci. China Chem. 56(2013) 595-603. DOI:10.1007/s11426-012-4812-6 |
| [28] |
G.I. Geertsema-Doornbusch, H.C. van der Mei, H.J. Busscher, J. Microbiol. Methods 18(1993) 61-68. DOI:10.1016/0167-7012(93)90072-P |
| [29] |
H.S. Courtney, I. Ofek, T. Penfound, et al., PloS ONE 4(2009) e4166. DOI:10.1371/journal.pone.0004166 |
| [30] |
A.Y. Bhagirath, Y. Li, D. Somayajula, et al., BMC Pulm. Med. 16(2016) 174-195. DOI:10.1186/s12890-016-0339-5 |
| [31] |
M.N. Hurley, M. Cámara, A.R. Smyth, Eur. Respir. J. 40(2012) 1014-1023. DOI:10.1183/09031936.00042012 |
| [32] |
A.M. Stevens, M. Schuster, K.P. Rumbaugh, J. Bacteriol. 194(2012) 2131-2141. DOI:10.1128/JB.00143-12 |
| [33] |
S.F. Bonté, T. Köhler, C. Van Delden, J. Antimicrob. Chemother. 52(2003) 598-604. DOI:10.1093/jac/dkg397 |
| [34] |
J.P. Pearson, K.M. Gray, L. Passador, et al., Proc. Natl. Acad. Sci. U. S. A. 91(1994) 197-201. |
| [35] |
J.P. Pearson, L. Passador, B.H. Iglewski, E.P. Greenberg, Proc. Natl. Acad. Sci. U. S. A. 92(1995) 1490-1494. DOI:10.1073/pnas.92.5.1490 |