 2018, Vol. 29
2018, Vol. 29 



b Institute for Molecules and Materials(IMM), Radboud University, 6525 AJ, Nijmegen, The Netherlands;
c Center for Photochemical Sciences, Bowling Green State University, Bowling Green OH 43403, USA;
d Solvay Specialty Polymers, Alpharetta GA 30005, USA;
e Australian Institute for Bioengineering and Nanotechnology(AIBN), The University of Queensland, Brisbane QLD 4072, Australia
Highly efficient nonlinear optical (NLO) materials play essential roles not only in advanced modern technologies ranging from super-resolution microscopy and lithography to terahertz (THz) generation, but also in many daily life aspects such as telecommunication, signal processing and data storage [1, 2]. In particular, NLO materials with well-defined architectures on the (sub)wavelength length scales are regarded as the key materials for developing the next generation integrated photonic circuits [3-5]. Second harmonic generation (SHG) [6] is one of the most widely studied and used second-order NLO effects, in which two photons of incident light combine and generate a photon with doubled frequency and energy. It is usually generated when an intense pulsed laser beam interacts with a medium that has a high NLO hyperpolarizability, β, and equal importantly, a non-centrosymmetric geometry [7]. Two-photon excited fluorescence (TPF), on the other hand, is a nonlinear optical up-conversion process described by a third-order nonlinear optical two-photon absorption (TPA) mechanism [8]. Despite that inorganic NLO materials such as barium borate (BBO), zinc oxide (ZnO), and potassium dihydrogen phosphate (KDP) are well-established and widely used, organic NLO materials have been demonstrated to have various advantages, including high hyperpolarizabilities, structure diversities and eases of process [9-11]. In particular, the tailor-made molecular structures of organic NLO dyes allow for hierarchical self-assembled architectures on the micro-and nano-scales by multiple supramolecular interactions [3, 12, 13]. This results in subwavelength scale materials with very well-defined structures and therefore highly anistropic optical properties, serving as ideal building blocks for integrated photonic circuit applications [12, 14-17].
The optimization of organic NLO materials usually involves the introduction of electron donating and accepting groups into a π-conjugated molecule [11, 18, 19]. By optimizing the chemical structures of the electron donors (D) and acceptors (A) as well as the π-bridges, intramolecular charge transfer (ICT) processes of these D-π-A molecular systems are facilitated and, therefore, manifested as a boost in their NLO hyperpolarizabilities [20, 21]. However, such an optimization in the molecular structure does not always lead to materials with high NLO susceptibility. Particularly for a second-order NLO process, a non-centrosymmetric organization of the dye molecules in these materials is essential. This is particularly challenging because these D-π-A compounds have a strong tendency to self-organize in centrosymmetric ways driven by the dipole-dipole interactions [22, 23]. Recently, we have found that fluorenone based compounds are very promising building blocks for multifunctional materials, exhibiting both second-and third-order NLO responses, integrated with distinctive waveguiding behaviours [24-26]. These fluorenone derivatives, despite of having a moderate ICT characteristic, favor synergistic non-covalent interactions such as hydrogen bonds and C-H…π interactions, leading to an accumulation of the molecular level NLO hyperpolarizabilities. The V-shaped geometry of fluorenone molecules results in different orientations of the transition dipole, μge, and the change of the permanent dipole from the ground state to the excited state, ∆μ, each playing a key role in the optimization of highly efficient NLO molecular materials based on fluorenone derivatives. In this context, further exploration of novel fluorenone based NLO dyes and a precise correlation between the optical responses with their molecular and supramolecular architectures is essential. Here, we have studied the polymorphs of a phenylethynyl functionalized fluorenone derivative, namely bis(2-phenylethyn-1-yl)fluoren-9-one (PAFO, Scheme S1 in Supporting information), and its controlled self-assembly in microstructures with different morphologies. These polymorphic microcrystals exhibit very distinctive nonlinear optical properties, which are highly dependent on their electronic and supramolecular structures, as revealed by the detailed NLO examinations in combination with theoretical calculations.
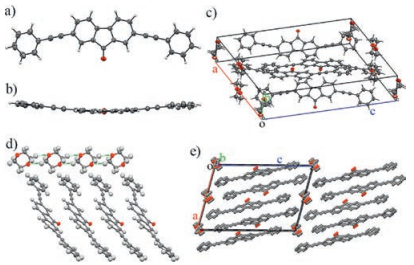
The synthesis of the title compound PAFO follows the reported procedures [27]. The compound is yellow and dissolves well (>5 mg/mL) in many common organic solvents such as toluene, dichloromethane (CH2Cl2), chloroform (CHCl3), tetrahydrofuran (THF), moderately (~ 0.5 mg/mL) in 1, 4-dioxane, and poorly (< 0.1 mg/mL) in highly polar solvents such as methanol (CH3OH), ethanol (EtOH), or acetonitrile (CH3CN). In this context, we are able to readily grow high quality single crystals from PAFO for X-ray diffraction (XRD) measurements via the liquid diffusion method. In a typical procedure, a concentrated solution of PAFO in CH2Cl2 (5 mg/mL, 2 mL) was gently covered with CH3CN (5 mL) with a clear boundary between the interfaces of the good and poor solvents. After the complete diffusion of the poor solvent CH3CN into the good solvent CH2Cl2, PAFO precipitated as needle-like crystals. Single crystal XRD analysis reveals that in these crystals (referred to as the α-phase), PAFO molecules are organized in an orthorhombic Pca21 space group, belonging to a non-centrosymmetric 222 point group, with unit cell parameters of a = 12.3209 (14) Å, b = 10.7613(13) Å, c = 29.421(3) Å, α = 90°, β = 90°, and γ = 90° (Table S1 in Supporting information). The compound has two molecular configurations in the unit cells (Figs. 1a and b). In one of the PAFO molecules, one of the phenyl rings is twisted from the fluorenone plane with a torsion angle of about 45.9°, while the other phenyl ring is nearly planar to the fluorenone moiety (torsion angle < 2°). In the other PAFO molecules, both of the phenyl rings are twisted from the fluorenone plane, but with very different torsion angles (45.0° and 14.2°, respectively). Such different configurations facilitate the intramolecular π-π stacking interaction, which serves together with the C—H‥π interactions and the dipole-dipole interactions as the supramolecular driving forces for the non-centrosymmetric molecular organizations (Figs. 1c and d).

|
Download:
|
| Fig. 1. Crystal structures of PAFO in the α-phase. a) The front view and b) the top view of the PAFO molecules with two different configurations. c) The unit cell of PAFO crystals in the α-phase. d) The packing diagram viewed from the crystallographic b-axis. | |
{kind=link}
If 1, 4-dioxane is chosen as the poor solvent for the liquid diffusion growth, belt-like single crystals of PAFO are formed after the complete mixing of the solvents. Single crystal XRD analysis suggests that these crystals actually belong to another crystal phase of PAFO (referred to as the β-phase), with a monoclinic P21/a space group (point group: 2/m) and unit cell parameters of a = 16.4740(13) Å, b = 5.8377(3) Å, c = 23.9662(13) Å, α = 90°, β = 104.803(5)°, and γ = 90° (Table S1). The PAFO molecules are in a bent configuration with a torsion angle of about 9.8° in the β-phase (Figs. 2a-c). The interactions between PAFO molecules are based on the similar π-π stacking and C—H‥π interactions as those in the α-phase. However, it is worth noting that the solvent molecules of 1, 4-dioxane are highly involved in defining the supramolecular organization of PAFO molecules in the β-phase crystals. The multiple hydrogen bonds between the 1, 4-dioxane molecules drive the formation of molecular chains of 1, 4-dioxane along the crystallographic b-axis (Fig. 2d) [28], which is also the direction along which the PAFO molecules are packed via π-π stacking interactions in a J-type aggregation (Fig. 2e).

|
Download:
|
| Fig. 2. Crystal structures of PAFO in the β-phase. a) The front view and b) the top view of the PAFO molecules with a bent configurations. c) The unit cell of PAFO crystals in the β-phase. d) The hydrogen bonded 1, 4-dioxane network for directing the π-π stacked molecular organization. e) The packing diagram viewed from the crystallographic b-axis. H atoms have been omitted for clarity. | |
{kind=link}
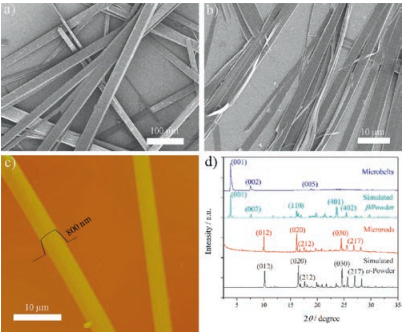
Well-defined micro-sized crystalline superstructures can be feasibly fabricated from the compound via simple solution based methods in a well-controlled manner. By using a re-precipitation method, in which the concentrated solution of PAFO in CH2Cl2 (5 mg/mL, 200 mL) was injected into 5 mL of CH3CN under vigorous stirring, the compound precipitated as microrod structures with widths of a few micrometers and lengths of hundreds of micrometers, as shown in the SEM image (Fig. 3a). If using a drop-casting method, in which the solution of PAFO in 1, 4-dioxane (0.2 mg/mL) was drop-casted onto the glass or silicon substrate, microbelt structures with widths of a few micrometers, lengths of hundreds of micrometers and thicknesses of a few hundred nanometers were observed after evaporation of the solvent (Figs. 3b and c). Powder XRD patterns of the as-prepared microstructures suggest that the microrods prepared by reprecipitation (solvents: CH2Cl2/CH3CN) belong to the α-phase polymorph of PAFO, while the microbelts prepared by drop-casting (solvent: 1, 4-dioxane) belong to the β-phase (Fig. 3d).

|
Download:
|
| Fig. 3. Morphology of the self-assembled microstructures of PAFO. The SEM images of a) microrods prepared by reprecipitation (solvents: CH2Cl2/CH3CN) and b) microbelts prepared by drop-casting (1, 4-dioxane). c) The AFM image of the asprepared microbelts. The cross-section shows a typical microbelt with a thickness of 800 nm. d) The XRD patterns of the as-prepared microstructures, in comparison with the simulated powder XRD of the α-and β-phase polymorphs, showing that the microrods and the microbelts belong to the α-and β-phase of the polymorphs of PAFO, respectively. | |
{kind=link}
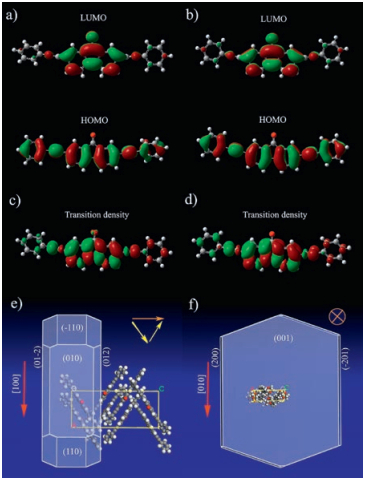
Theoretical calculations were carried out to gain more insights into the electronic and supramolecular structures of these microsized self-assemblies. The electronic structures of PAFO in both the α-and β-phases in the ground and excited states have been evaluated at the level of B3LYP/6-31g**. The shapes of the frontier highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs), as shown in Figs. 4a and b, and Fig. S2 in Supporting information, indicate clear charge transfer in PAFO in both phases. The transition dipoles corresponding to the S0-S1 transitions of PAFO in both the α-and β-phases, as shown in their respective transition densities (Figs. 4c and d, and Fig. S2), are mainly along the direction of the molecular long axes of PAFO molecules. As calculated by the Morphology module of the Material Studio package, in the a-phase, the microrod is grown along the [100] direction of the crystal (the π-π stacking direction). In this context, the overall transition dipole summed up from those of the "zig-zag" oriented individual molecules, is along the [010] direction, which is perpendicular to the long axis of the microrods (Fig. 4e). The β-phase microbelt however is grown along the [010] direction of the crystal (Fig. 4f), which is the direction of the hydrogen bonded molecular chains of the solvent 1, 4-dioxane [28]. Therefore, the transition dipole, being parallel to the molecular long axis (the crystallographic a-axis), is oriented perpendicular to the big-area surface, namely the (001) face of the microbelts.

|
Download:
|
| Fig. 4. Simulated electronic and supramolecular structures of the self-assembled polymorphic microcrystals. The calculated HOMOs and LUMOs of PAFO in the a) α-phase and b) the β-phase. The transition densities corresponding to the S0-S1 transition of PAFO in c) the α-phase and d) the β-phase. Predicted morphologies of PAFO in e) the α-phase and f) the β-phase. The size and color of the spheres in panels a), b), c) and d) represent the amplitude and sign (red is negative and green is positive) of the charge, respectively. The red arrows in panels e) and f) represent the growth directions of the microstructures; the yellow arrows show the orientations of the transition dipole of the individual molecules while the orange arrows show the overall transition dipoles in the microcrystals. | |
{kind=link}
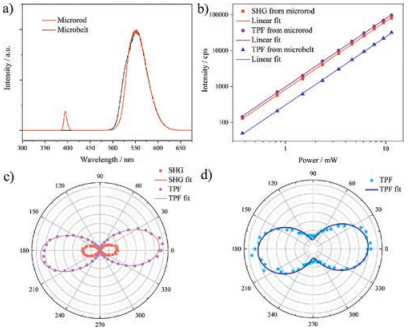
With the clear pictures of the (supra)molecular and electronic structures of both microstructures as discussed above, we have studied the NLO properties of the single crystals of the microrod and microbelt morphologies using a home-built laser scanning optical microscope with a femtosecond laser pump at 800 nm (120 fs, 82 MHz) [24]. The incidence and detection angles were both set at 45°, and the one dimensional (1D) microrod and microbelt were placed perpendicular to the plane of incidence. As shown in the NLO spectra (Fig. 5a), both the α-phase microrod and β-phase microbelt exhibited strong TPF signals with a very similar spectral feature peaking at about 550 nm. Meanwhile, one can clearly notice a pronounced SHG signal at 400 nm in the spectrum of the microrod, as expected from the non-centrosymmetric structure of the α-phase crystals with the point group of 222 (space group: Pca21). The intensities of all these NLO signals from both the microrod and microbelt scaled quadratically with the incident power, demonstrating the two-photon nature of both SHG and TPF (Fig. 5b). The polarization dependencies of the NLO signals of the 1D microstructures were studied relative to their well-defined supramolecular and electronic structures. The results, as shown in Figs. 5c and d, demonstrated that the SHG and TPF from both 1D microstructures exhibited the strongest signals when the excitation beam was p-polarized, which is perpendicular to the vertically oriented microrod and microbelt. This is in line with the welloriented transition dipoles of the microstructures of both the α-and β-polymorphs of PAFO. The polarization ratio, ρ = (Imax-Imin)/(Imax + Imin), was determined to be 0.998 ± 0.001 and 0.938 ± 0.005 for the SHG and TPF of the microrod, respectively, in comparison to 0.694 ± 0.003 of that of the TPF of the microbelt. The very high polarization ratios of both SHG and TPF from the microrod are attributed to its well-defined supramolecular structure which leads to the very well-oriented transition dipole moments. The efficiency of the second-order nonlinearity of the PAFO microrods was evaluated by measuring their SHG intensity compared with that of inorganic material Y-cut quartz under the same measurement conditions. The results suggest that the PAFO microrods have a relative value of about 7.95 and 0.54 times to that of the Y-cut quartz at 740 nm and 800 nm, respectively.

|
Download:
|
| Fig. 5. Nonlinear optical responses of the self-assembled microstructures of PAFO. a) NLO spectra. b) The logarithmic plots of the power dependence of SHG and TPF signals. The slopes of their linear fits are 2.0 ± 0.1. The polarization dependence plots of the NLO signals from the c) microrod and d) microbelt, respectively. The dots are experimental data and the solid lines are the cos4θ and cos2θ fits for SHG and TPF, respectively [24]. Both the microrod and microbelt are vertically aligned relative to the plane of incidence. The incidence pump was p-polarized for panels a) and b), and the power was 2.2 mW for panels a), c) and d). The pump wavelength was 800 nm. | |
{kind=link}
In summary, we have studied the self-assembly of the polymorphic microstructures with different morphologies from a phenylethynyl functionalized fluorenone derivative PAFO, and investigated their distinctive nonlinear optical properties. The observed second-and third-order NLO responses of these microstructures are highly anisotropic, depending on their welldefined supramolecular and electronic structures. Our study highlights the importance of the supramolecular organizations for future design of molecular photonic materials. The dual NLO responses of the microstructures along with the very high polarization ratios promise the possible applications of these materials in future photonic circuits.
AcknowledgmentsThe authors are grateful for the financial supports from the National Natural Science Foundation of China (NSFC) (Nos.21773168, 51503143 and 21761132007), the Tianjin Natural Science Foundation (No. 16JCQNJC05000), the Innovation Foundation of Tianjin University (No. 2016XRX-0017), the China International Science and Technology Projects (No. S2016G3413), The Netherlands Organization for Scientific Research (NWO) with the Veni Grant (No. 680-47-437), The Royal Netherlands Academy of Arts and Sciences (KNAW) with the China-Exchange Program (No. 530-4CDP02), the Tianjin 1000 Youth Talents Plan, and the Chinese Scholarship Council (CSC). Technical assistances from Dr. E. Pierson and G.J. Janssen from the General Instruments (GI) Department, A. Toonen from SSI Department of Radboud University are highly appreciated.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2017.11.001.
| [1] |
E. Garmire, Opt. Express 21(2013) 30532-30544. DOI:10.1364/OE.21.030532 |
| [2] |
J. Xu, R.W. Boyd, G.L. Fischer, Nonlinear Optical Materials, Reference Module, Materials Science and Materials Engineering, Elsevier B.V.(2016), pp. 6237-6244. |
| [3] |
J. Xu, S. Semin, T. Rasing, A.E. Rowan, Small 11(2015) 1113-1129. DOI:10.1002/smll.v11.9-10 |
| [4] |
R. Yan, D. Gargas, P. Yang, Nature Photon. 3(2009) 569-576. DOI:10.1038/nphoton.2009.184 |
| [5] |
T. Chervy, J. Xu, Y. Duan, et al., Nano Lett. 16(2016) 7352-7356. DOI:10.1021/acs.nanolett.6b02567 |
| [6] |
P.A. Franken, G. Weinreich, C.W. Peters, A.E. Hill, Phys. Rev. Lett. 7(1961) 118-119. DOI:10.1103/PhysRevLett.7.118 |
| [7] |
S.R. Marder, J.E. Sohn, G.D. Stucky, Materials for Nonlinear Optics:Chemical Perspectives, American Chemical Society, Boston(1991). |
| [8] |
G.S. He, L.S. Tan, Q. Zheng, P.N. Prasad, Chem. Rev. 108(2008) 1245-1330. DOI:10.1021/cr050054x |
| [9] |
C. Wang, C. Fan, C. Yuan, et al., RSC Adv. 7(2017) 4825-4829. DOI:10.1039/C6RA25408J |
| [10] |
M. de Torres, S. Semin, I. Razdolski, et al., Chem. Commun. 51(2015) 2855-2858. DOI:10.1039/C4CC09592H |
| [11] |
J.X. Dong, H.L. Zhang, Chin. Chem. Lett. 27(2016) 1097-1104. DOI:10.1016/j.cclet.2016.05.005 |
| [12] | |
| [13] |
H.B. Liu, J. Xu, Y.J. Li, Y.L. Li, Acc. Chem. Res. 43(2010) 1496-1508. DOI:10.1021/ar100084y |
| [14] |
C. Zhang, Y.L. Yan, Y.S. Zhao, J.N. Yao, Acc. Chem. Res. 47(2014) 3448-3458. DOI:10.1021/ar500192v |
| [15] |
D.P. Yan, D.G. Evans, Mater. Horiz. 1(2014) 46-57. DOI:10.1039/C3MH00023K |
| [16] |
Y. Luo, C. Yuan, J. Xu, et al., ACS Appl. Mater. Inter. 9(2017) 30862-30871. DOI:10.1021/acsami.7b10109 |
| [17] |
B. Tang, Z. Zhang, H. Liu, H. Zhang, Chin. Chem. Lett. (2017) https://doi.org/10.1016/j.cclet.2017.08.045.
|
| [18] |
J.E. Reeve, H.L. Anderson, K. Clays, Phys. Chem. Chem. Phys. 12(2010) 13484-13498. DOI:10.1039/c003720f |
| [19] |
Z.B. Sun, S.Y. Li, Z.Q. Liu, C.H. Zhao, Chin. Chem. Lett. 27(2016) 1131-1138. DOI:10.1016/j.cclet.2016.06.007 |
| [20] |
Y.J. Li, T.F. Liu, H.B. Liu, M.Z. Tian, Y.L. Li, Acc. Chem. Res. 47(2014) 1186-1198. DOI:10.1021/ar400264e |
| [21] |
M. Kivala, F. Diederich, Acc. Chem. Res. 42(2009) 235-248. DOI:10.1021/ar8001238 |
| [22] |
J. Xu, L.P. Wen, W.D. Zhou, et al., J. Phys. Chem. C 113(2009) 5924-5932. DOI:10.1021/jp809258h |
| [23] |
J. Xu, X.F. Liu, J. Lv, et al., Langmuir 24(2008) 4231-4237. DOI:10.1021/la703662w |
| [24] |
J. Xu, S. Semin, D. Niedzialek, et al., Adv. Mater. 25(2013) 2084-2089. DOI:10.1002/adma.201204237 |
| [25] |
J. Xu, S. Semin, J. Cremers, L. Wang, et al., Adv. Opt. Mater. 3(2015) 948-956. DOI:10.1002/adom.201400637 |
| [26] |
Y. Duan, C. Ju, G. Yang, et al., Adv. Funct. Mater. 26(2016) 8968-8977. DOI:10.1002/adfm.v26.48 |
| [27] |
L.A. Estrada, J.E. Yarnell, D.C. Neckers, J. Phys, Chem. A 115(2011) 6366-6375. |
| [28] |
J. Xu, H.Y. Zheng, H.B. Liu, et al., J. Phys. Chem. C 114(2010) 2925-2931. DOI:10.1021/jp911595m |