 2018, Vol. 29
2018, Vol. 29 



b Laboratory for Photonics and Interfaces, School of Basic Sciences, Swiss Federal Institute of Technology, Lausanne 1015, Switzerland
Dye-sensitized solar cell (DSSC), since it was firstly reported in 1991 by O'Reagan and Grätzel [1], initiated the possibility of maximizing the harnessing of solar light in a cost-efficient way. The principle of the DSSC is based on a broadband inorganic semiconductor scaffold, e.g., TiO2, which is sensitized with a strongly absorbing dye. High efficiencies up to 13% have been achieved by employing a meso-substituted porphyrin dye in conjunction with a tris(2, 2'-bipyridine)cobalt(Ⅱ/Ⅲ)redox couple [2]. Recently, Hanaya et al. [3-5] developed a collaborative sensitization strategy combining ADEKA-1 and LEG4 dyes in DSSC with silyl-anchor and carboxy-anchor, respectively. They realized a high light-to-electric power conversion efficiency (PCE) of over 14.5% under one sun illumination due to successful pinhole filling by dye layers with multiple coadsorbates to reduce interfacial charge recombination.
Among the five key components of DSSC: Transparent conductive oxide such as fluorine-doped SnO2 (FTO), mesoporous semiconductor metal oxide (such as nanocrystalline TiO2), sensitizer (dye), electrolyte/hole transporter, and counter electrode (platinum or carbon on FTO), the dye takes up a big fraction of the total cost. Despite the excellent light harvesting ability, the tedious synthesis of porphyrin dyes labels itself as a luxurious choice for DSSC community. In the meantime, the easily accessible metal-free organic dyes based on arylamines especially triphenylamine (TPA) (Fig. 1) as electron donating fragments have shown excellent electronic properties [6-14].

|
Download:
|
| Fig. 1. Comparison of triphenylamine and various fused-acenes (structures and DFT calculated highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO)). | |
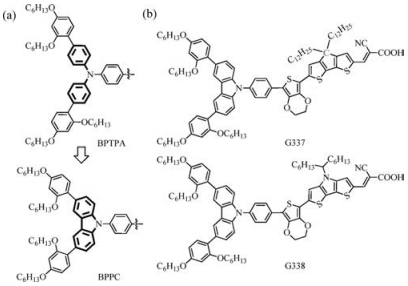
On the other hand, organic dyes based on fused TPA, such as 9-phenyl carbazole donors, have also been subjects of investigations over the last decade [15-18]. This could be explained by its interesting features such as low cost of the starting material (9Hcarbazole), good chemical and environmental stability provided by the fully aromatic unit, easy substitution of the nitrogen atom or the aromatic rings with a wide range of functional groups permitting a better solubility and a fine tuning of the electronic and optical properties. At the same time, a more directional charge separation can be found in the molecular orbital (MO) of 9-phenyl carbazole than TPA based on density function theory (DFT) simulation (Fig. 1). The effect of ring fusion can also be observed in the choosing of -bridge units.
The most successful example can be found in the work by Wang et al. [19] that by employing 4, 4-dihexyl-4H-cyclopenta[2, 1-b:3, 4-b']dithiophene (CPDT) instead of the prevailing 2, 2'-dithiophene (DT) as the building block they got extremely high-molarabsorption-coefficient organic sensitizer C218. Dithieno[3, 2-b:2', 3'-d]pyrrole (DTP) has also been evaluated as a promising p-bridge in sensitizers [20]. The calculated MO shows that a bigger node is shown on the N atom of DTP in LUMO orbital of than that of CPDT although they have very similar HOMO orbitals (Fig. 1). In this work, we designed two new dyes combining substituted carbazole (3, 6-bis(2, 4-bis(hexyloxy)phenyl)-9-phenyl-9H-carbazole, BPPC) as donor moiety mimicking the famous (N-(2', 4'-bis(hexyloxy)-[1, 1'-biphenyl]-4-yl)-N-(4-bromophenyl)-2', 4'-bis(hexyloxy)-[1, 1'-biphenyl]-4-amine) (BPTPA) [7] to reduce charge recombination in the device by preventing the oxidized redox mediator to access the TiO2 surface (Fig. 2a) and two kinds of fused acenes (CPDT and DTP) as the p-bridges to evaluate their compatibility with cobalt-based electrolyte. Additionally, 3, 4-ethoxylene dioxythiophene (EDOT) is introduced into sensitizers to broaden the spectral response [21] (Fig. 2b). The DSSCs using these dyes and ([CoⅡ/Ⅲ(bpy)3]2+/3+ redox mediator (tris(2, 2'-bipyridine)cobalt(Ⅱ) and tris(2, 2'-bipyridine) cobalt(Ⅲ) complexes) were characterized by transient absorption spectroscopy, and transient photovoltage decay, revealing the potential of DTP as a bridge in the design of sensitizers and efficiency up to 7.45%.

|
Download:
|
| Fig. 2. Structures of (a) evolution from BPTPA to BPPC, (b) G337 and G338. | |
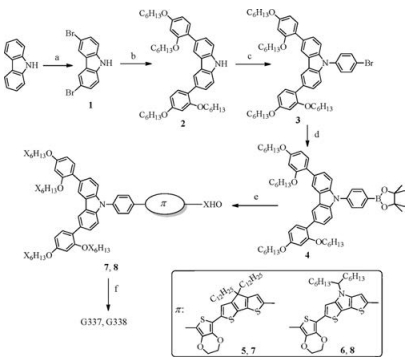
The synthetic pathways that lead to the formation of the targeted dyes are shown in Scheme 1. Detailed synthetic routes to the intermediate compounds 7 are described in Supporting information. Carbazole-substituted donor moiety 4 was synthesized according to a four-step synthesis as illustrated in Scheme 1 in an overall yield of 50%. The differences between 4 and previously reported BPTPA donor is the donor moiety in which the diphenylamine is fused to form a carbazole structure. Palladium-catalyzed Suzuki coupling of 4 with bridging units 5 and 6 respectively gave aldehydes 7 and 8 in 65% and 60% isolated yield. A Knoevenagel condensation of the aldehydes with cyanoacetic acid, in the presence of piperidine, afforded the title sensitizers G337 and G338 in 45% and 47% yield, respectively. These target sensitizer dyes were well characterized by 1H NMR, 13C NMR, and HRMS.

|
Download:
|
| Scheme 1. Synthesis of dyes G337 and G338. Reagents and conditions: (a) NBS, DMF, r.t.; (b) Bis(hexyloxy)phenyl)-tetramethyl-dioxaborolane, K2CO3, Pd(0); (c) K3PO4, CuI, 110 ℃; (d) n-BuLi, 2-isobutyl-4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane, THF; (e) Pd(0), toluene, 2 mol/L K2CO3, reflux; (f) Cyano-acetic acid, piperidine, CHCl3, reflux. | |
The optical properties of G337 and G338 are strongly solventand pH-dependent (Fig. 3 and Table 1) due to changes in the protonation state of the cyano-acrylic acceptor. The absorption spectra in dichloromethane solution exhibit maxima at 548 nm and 544 nm, respectively (Fig. 3a and Table 1, and Table S1 in Supporting information). With 3, 6-carbazole as donor group, dyes G337 and G338 exhibit higher molar extinction coefficients than the dyes with the same bridge but TPA as donor groups, owing to their electron-rich and rigid 2, 7-disubstituted carbazole framework [6], which enhances the transition probability for both the π-π* and charge-transfer transitions. In particular, the absorption maxima of G338 is blue-shifted by 4 nm compared to that of G337. The unexpected hypsochromic shift observed for the DTP relative to the CPDT dyes indicates that the nitrogen bridge weakens charge transfer effect, which confirms the intramolecular charge transfer (ICT) character of the main transition.

|
Download:
|
| Fig. 3. (a) Steady-state absorption in dichloromethane solution. Solvatochromic effect in the absorption of (b) G337 and (c) G338 in various solvents. | |
|
|
Table 1 Photophysical and electrochemical properties of the dyes. |

Both dyes showed one significant charge-transfer transition around 550 nm. To further confirm the charge-transfer nature of the low-energy absorption band, absorption spectra of the dyes were measured in solvents of different polarity. The absorption curves were illustrated in Figs. 3b and c and the corresponding data are compiled in Table S1. Both dyes showed negative solvatochromism for the longer-wavelength absorption band. The most red-shifted absorption was observed in dichloromethane and the absorption peak shifted to shorter wavelength on increasing the polarity. This result suggests an effective solvation of the dyes with increasing solvent polarity. The blue-shift in polar solvents may be induced by the acid-base equilibrium that is exhibited by the dyes. We speculate that the carboxylic-acid unit is partially deprotonated in polar solvents that are capable of abstracting the acidic hydrogen atom, either by hydrogen bonding or owing to the basic nature of the solvent. This abstraction may, in turn, impede the donor-acceptor interactions in the dye and produce a hypsochromic shift of the charge-transfer transition [22-24]. The absorption spectra of the dyes in N, N-dimethylformamide (DMF) displayed a prominent blue-shift for the charge-transfer transition compared to those in other solvents, which was consistent with the basicity of DMF.
The energy levels of the dyes with respect to the electrochemical potential of the electrolyte and conduction band of TiO2 (-0.5 V vs. NHE) [25] also play an important role in the resulting DSSC performance. The ground state oxidation potential (Eox) of G337 and G338 are measured at 0.83 V and 0.91 V vs. NHE [26]. These values could provide ample driving force for regeneration from a [CoⅡ/Ⅲ(bpy)3]2+/3+ electrolyte (0.56 V vs. NHE). Using the Eox of the dyes and the estimated E0-0 value, the excited-state oxidation potentials (ES+/S*) for the carbazole donor dyes were estimated at -1.15 and -1.14 V, respectively. The driving force for injection of the photoexcited electron into the TiO2 conduction band is similar to their TPA analogs, thus ensuring quantitative injection quantum yields into TiO2 [6].
In order to gain further insight into the photophysical properties, theoretical calculations of molecular energy levels and orbital distributions was carried out based on DFT and the results are illustrated in Fig. S1 (Supporting information). In the ground state, the electron density of the HOMO in the dyes is mainly populated over the carbazole donor and EDOT unit with few contributions from the latter bridging blocks, whereas the LUMO orbital is delocalized through the EDOT, CPDT (DTP) fragments, and finally on cyanoacrylic acid groups. This spatially directed separation of the frontier orbitals strongly promotes intramolecular charge separation, hence favoring efficient electron injection from the excited state of the dye into the semiconducting oxide, and limiting charge recombination processes. In addition, the hole localization on the carbazole fragment facilitates the electron donor to approach, promoting the fast dye regeneration.
We performed transient absorption (TA) measurements and estimated the time constants of charge recombination and dye regeneration (Fig. 4 and Fig. S2 in Supporting information). Following excitation at 470 nm, the oxidized forms of the dyes have absorption spectra that sharply contrast with their neutral counterparts. The signals that appear at ca. 600-1000 nm are characteristic of TA of 9-phenylcarbazole radicals. Time constants are derived from TA decays of the corresponding radical. The signals were fitted with a single exponential component decay, ∆A (t)∝A0exp[-(t/τ)], where A0 is the pre-exponential factor and τ is the characteristic time [27]. The fits are represented as solid lines in Figs. 4a and b, with data reported in Table 2. In the absence of electrolyte (dummy cells in acetonitrile), the time constants τrec of G338 is higher than that of G337, indicating a slower recombination rate between electrons injected into the TiO2 and the oxidized form of the G338. The lower τrec value measured for G337 suggests that charges could recombine slightly faster when the CPDT bridge is used. In the presence of active cobalt(Ⅱ/Ⅲ) electrolyte, the TA decays were accelerated, with τreg values of 95 ms and 30 ms for G337 and G338, respectively. The DTP bridge appears more favorable for the regeneration of the oxidized dye than CPDT case according to its short τreg. Also in the case of G338, the τreg/τrec ratio is much larger, suggesting extremely favorable chargetransfer kinetics compared to the other system, which results in nearly quantitative regeneration quantum yields

|
Download:
|
| Fig. 4. TA decay traces of 8.0 mm thick TiO2 films sensitized with G337 (a) and G338 (b). Decays in inert (black) and [CoⅡ/Ⅲ(bpy)3]2+/3+ (red) electrolytes. Excited with a laser pulse of 50 mJ/cm2 (λexc = 530 nm), probe at 750 nm. | |
|
|
Table 2 Detailed photovoltaic parameters.a |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The incident photon-to-current efficiency (IPCE) spectra were measured on films coated with G337 and G338 with an optimized [CoⅡ/Ⅲ(bpy)3]2+/3+ electrolyte composition. The IPCEs exhibit a similar shape for the two dyes with intensity maxima around 60%-80% (Fig. 5a). A blue-shift of 30 nm in the onset is observed for the given DTP dye (G338) compared to its CPDT analog (G337), which is in agreement with their estimated E0-0 values.

|
Download:
|
| Fig. 5. (a) IPCE spectra of the sensitizers and (b) Photovoltaic performance obtained at 100 mW/cm2 AM 1.5G illumination. (c) Electron lifetimes and (d) changes in VOC as measured by transient photovoltage and photocurrent decay respectively. | |
{kind=link}
The PCEs of these DSSCs were evaluated by recording the J-V characteristics under simulated AM 1.5G illumination (Fig. 5b). The detailed output parameters of short-circuit current density (JSC), open-circuit voltage (VOC), fill factor (FF), and PCEs (η) are summarized in Table 2. Unexpectedly, both the photocurrent densities (JSC) and open circuit voltage (VOC) measured for the G338 are higher despite the low IPCE. The higher VOC from the device based on G338 arises from the low recombination rate according to the TA measurement. The low JSC of G337 contrasting to the high IPCE response can be explained by its larger regeneration time scale than G338 [28]. In the meantime, the two bulky dodecyl chains in G337 decreased the loading of the dye molecules on the surface, which additionally contributes to the low JSC of G337.
To further provide a rationale for the origin of the different VOC, the dependence of TiO2 electron lifetimes on capacitance as recorded from photocurrent decay measurements for the investigated dyes was measured (Fig. 5c). The use of DTP instead of CPDT gave a device with longer electron lifetimes, indicating that the recombination reaction between the electrons on the TiO2 surface and the trivalent cobalt complex in the presence of dyes with CPDT as the bridge was considerably faster than that with DTP. The dye with DTP bridge also exhibited relatively lower dark current density (Fig. S3 in Supporting information). As can be seen from Fig. 5d, similar to our previous result [29], the introduction of CPDT bridge lowers the conduction band of TiO2 (EF) more significantly, which will further depress the VOC of G337 based devices.
To conclude, we developed two new sensitizers with three kinds of fused acenes as both donor and π-bridge groups. The molecularly designed donor group provide compatibility with cobalt-based redox shuttles. Both the transient absorption spectroscopy and transient photovoltage decay revealed the potential of DTP dyes in providing a large VOC (~850 mV). This feature can overcome the nettlesome phenomenon that large VOC values are usually obtained with high oxidation redox mediators, which is detrimental to dye regeneration and current densities. A promising PCE of 7.45% is obtained with G338 and further work is currently underway to improve the spectral response of such DTP dyes.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2017.09.056.
| [1] |
B. O'Regan, M. Grätzel, Nature 353(1991) 737-740. DOI:10.1038/353737a0 |
| [2] |
S. Mathew, A. Yella, P. Gao, et al., Nat. Chem. 6(2014) 242-247. DOI:10.1038/nchem.1861 |
| [3] |
K. Kakiage, Y. Aoyama, T. Yano, et al., Chem. Commun. 50(2014) 6379-6381. DOI:10.1039/c4cc02192d |
| [4] |
K. Kakiage, Y. Aoyama, T. Yano, et al., Chem. Commun. 51(2015) 6315-6317. DOI:10.1039/C5CC00464K |
| [5] |
K. Kakiage, Y. Aoyama, T. Yano, et al., Chem. Commun. 51(2015) 15894-15897. DOI:10.1039/C5CC06759F |
| [6] |
A. Kaeser, B. Delavaux-Nicot, C. Duhayon, Y. Coppel, J.F. Nierengarten, Inorg. Chem. 52(2013) 14343-14354. DOI:10.1021/ic402342y |
| [7] |
P. Gao, Y.J. Kim, J.H. Yum, et al., J. Mater. Chem. A 1(2013) 5535-5544. DOI:10.1039/c3ta10632b |
| [8] |
P. Gao, H.N. Tsao, M. Grätzel, M.K. Nazeeruddin, Org. Lett. 14(2012) 4330-4333. DOI:10.1021/ol301730c |
| [9] |
A. Yella, R. Humphry-Baker, B.F.E. Curchod, et al., Chem. Mater. 25(2013) 2733-2739. DOI:10.1021/cm401593b |
| [10] |
N. Cai, Y. Wang, M. Xu, et al., Adv. Funct. Mater. 23(2013) 1846-1854. DOI:10.1002/adfm.v23.14 |
| [11] |
M. Zhang, J. Zhang, Y. Fan, et al., Energy Environ. Sci. 6(2013) 2939-2943. DOI:10.1039/c3ee42431f |
| [12] |
Y. Xie, L. Han, C.S. Ge, Y.H. Cui, J.R. Gao, Chin. Chem. Lett. 28(2017) 285-292. DOI:10.1016/j.cclet.2016.06.042 |
| [13] |
Y. Wu, X. Li, Chin. Chem. Lett. 27(2016) 927-932. DOI:10.1016/j.cclet.2016.04.010 |
| [14] |
J.S. Luo, Z.Q. Wan, C.Y. Jia, Chin. Chem. Lett. 27(2016) 1304-1318. DOI:10.1016/j.cclet.2016.07.002 |
| [15] |
N. Koumura, Z.S. Wang, S. Mori, et al., J. Am. Chem. Soc. 128(2006) 14256-14257. DOI:10.1021/ja0645640 |
| [16] |
N. Koumura, Z.S. Wang, M. Miyashita, et al., J. Mater. Chem. 19(2009) 4829-4836. DOI:10.1039/b905831a |
| [17] |
K. Hara, Z.S. Wang, Y. Cui, A. Furube, N. Koumura, Energy Environ. Sci. 2(2009) 1109. DOI:10.1039/b907486d |
| [18] |
M. Liang, J. Chen, Chem. Soc. Rev. 42(2013) 3453-3488. DOI:10.1039/c3cs35372a |
| [19] |
R. Li, J. Liu, N. Cai, M. Zhang, P. Wang, J. Phys. Chem. B 114(2010) 4461-4464. DOI:10.1021/jp101222s |
| [20] |
L.E. Polander, A. Yella, J. Teuscher, et al., Chem. Mater. 25(2013) 2642-2648. DOI:10.1021/cm401144j |
| [21] |
M.W. Lee, J.Y. Kim, D.H. Lee, M.J. Ko, ACS Appl. Mater. Interfaces 6(2014) 4102-4108. DOI:10.1021/am405686z |
| [22] |
Q. Feng, X. Lu, G. Zhou, Z.S. Wang, Phys. Chem. Chem. Phys. 14(2012) 7993-7999. DOI:10.1039/c2cp40872d |
| [23] |
E. Miyazaki, T. Okanishi, Y. Suzuki, et al., Bull. Chem. Soc. Jpn. 84(2011) 459-465. DOI:10.1246/bcsj.20100302 |
| [24] |
A. Baheti, P. Tyagi, K.R.J. Thomas, Y.C. Hsu, J.T. Lin, J. Phys. Chem. C 113(2009) 8541-8547. DOI:10.1021/jp902206g |
| [25] |
A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo, H. Pettersson, Chem. Rev. 110(2010) 6595-6663. DOI:10.1021/cr900356p |
| [26] |
N.G. Connelly, W.E. Geiger, Chem. Rev. 96(1996) 877-910. DOI:10.1021/cr940053x |
| [27] |
A.Y. Anderson, P.R.F. Barnes, J.R. Durrant, O'Regan B.C., J. Phys. Chem. C 115(2011) 2439-2447. DOI:10.1021/jp1101048 |
| [28] |
S. Aghazada, P. Gao, A. Yella, et al., Inorg. Chem. 55(2016) 6653-6659. DOI:10.1021/acs.inorgchem.6b00842 |
| [29] |
P. Gao, H.N. Tsao, C. Yi, M. Grätzel, M.K. Nazeeruddin, Adv. Energy Mater. 4(2014) 1301485. DOI:10.1002/aenm.201301485 |