 2018, Vol. 29
2018, Vol. 29 


 ,
Paula de Almeida,
Onno ven den Boomen,
Zaskia H. Eksteen-Akeroyd,
Roel Hammink,
Maarten Jaspers,
Stijn Kragt,
Mathijs F.J. Mabesoone,
Roeland J.M. Nolte,
Alan E. Rowan,
Martin G.T.A. Rutten,
Vincent A.A. Le Sage,
Daniël C. Schoenmakers,
Chengfen Xing,
Jialiang Xu
,
Paula de Almeida,
Onno ven den Boomen,
Zaskia H. Eksteen-Akeroyd,
Roel Hammink,
Maarten Jaspers,
Stijn Kragt,
Mathijs F.J. Mabesoone,
Roeland J.M. Nolte,
Alan E. Rowan,
Martin G.T.A. Rutten,
Vincent A.A. Le Sage,
Daniël C. Schoenmakers,
Chengfen Xing,
Jialiang Xu
Thermoresponsive polymer solutions show an abrupt transition from a dissolved to a precipitated state or gel. Many examples in the literature describe polymers with a lower critical solution temperature (LCST) when dissolved in water [1]. At low temperatures, such a polymer is hydrated by the solvent, often involving hydrogen-bonding interactions with the solvent, and forms a homogeneous solution. Beyond the LCST, however, intramolecular interactions are favoured and the polymer chain conformation changes from an extended coil to a collapsed globular conformation, which often leads to precipitation of the polymer from the solution. The potential applications of thermosensitive polymers are endless, ranging, for instance, from drug delivery and biomedical applications [2] (including polymers with transient thermal properties [3]) to smart functional surfaces [4].
A key parameter in LCST polymers is the transition temperature, which besides the molecular structure may depend on the polymer concentration, its molecular weight and the presence of salts or other solutes in the solution. The most studied material, poly(Nisopropylacrylamide) or pNiPAM shows an LCST of 32 ℃, close to body temperature [5]. Over the years, many differently substituted poly(meth)acrylamides and other polymers have been characterised and transition temperatures virtually anywhere between 10 ℃ and 90 ℃ have been found [1]. This list also includes semi-biological materials, for instance elastin-like polypeptides [6] and cellulose derivatives [7]. A proficient approach to precisely tailor the LCST of a polymer solution is to prepare copolymers [8]. The copolymer LCST usually falls between the transition temperatures of the homopolymers and the copolymer ratio determines the exact value.
A different class of LCST polymers have small ethylene glycol side chains or tails; the main chain may vary wildly from poly(vinyl ether)s [9] and poly(meth)acrylates [10, 11] to polyphosphazenes [12] and even polynorbornenes [13] and polyisocyanates [14]. The LCST behaviour of this class of polymer materials is governed by the small ethylene glycol (EG) group, and as such, the transition temperature is determined by the EG length, as well as its endgroup (usually methyl). Work on EG-substituted polymethacrylates [15] showed that also in these materials, tight control of the transition temperature is feasible by copolymerising monomers with EG tails of different lengths.
When LCST monomers are copolymerised with small amounts of crosslinkers, covalently linked hydrogels are formed. Heating beyond the transition temperature causes a collapse of the hydrogel network usually accompanied by significant syneresis. Since the transition is fully reversible, cooling leads to reswelling of the gel to its former state. Recently, our group developed a hydrogel based on semi-flexible polyisocyanides, decorated with EG groups (PIC, Fig. 1) [16]. The EG substituents render the aqueous polymer solutions thermoresponsive. Because of their rigid conformation and high molecular weight, the polymers do not precipitate, but rather form a network of bundled polymer chains that is capable of holding water over 10, 000 × its own weight [17]. The bundled structure of relatively stiff polymer chains causes the PIC gels to become much stiffer under stress [16, 18]. This process, termed strain-stiffening is instantaneous and fully reversible. The transition into the strain-stiffening regime occurs for PIC gels already at very low stresses, in line with other biological gels, such as actin, fibrin and collagen [19]. The close structural and mechanical resemblance with these biopolymer gels is unique for synthetic hydrogels and forms the basis for applications in the biomedical field, for instance in tissue engineering [20].

|
Download:
|
| Fig. 1. General structure of PIC random co-polymers. (a) PIC monomers with different ethylene glycol tail lengths (a = 2-4, b = 4 or 8–12) are randomly copolymerised. (b) Schematic drawing of the structure of the helical polymer chain with two different length EG tails incorporated. The main chain is wellorganised while the EG groups are expected to have a more disordered arrangement. | |
For PIC gels and many other hydrogels, the LCST is closely related to the mechanical properties and as such, crucial for any application. In this paper, we show how we can manipulate the LCST by varying the length of the grafted EG tails. Copolymerisation of different monomers (Fig. 1) allows us to carefully fine-tune the transition temperatures of PIC hydrogels.
Homopolymers and copolymers were synthesised from a series of isocyanide monomers with different EG tail lengths (Scheme 1), following literature protocols [21]. For EG tails longer than 4, a commercial mixture of EG monomethyl ether HO(CH2CH2O)bMe with b = 8-12 was used as starting material in the monomer synthesis; MALDI-ToF mass spectroscopy shows a peak at b = 11. In addition, all of our monomers have methyl end-groups. The endgroup has a large effect on the transition temperature of the polymer [22]. For the formation of the copolymers, the appropriate monomers were dissolved in toluene at the desired molar ratio (see Table 1). To this solution, a catalyst solution of nickel(Ⅱ) perchlorate was added and the mixture was allowed to react for one or two days, until FT-IR indicated complete consumption of the monomer (as indicated by the disappearance of the characteristic isocyanide absorbance at 2140 cm-1). After the reaction, pure polymers were obtained by several precipitations (details are given in Supporting information).

|
Download:
|
| Scheme 1. Copolymerisation reaction and polymers measured. Homopolymers were prepared from isocyanides with a = 3 or 4. Co-polymers (a, b = 2, 3, 4 or 8–12) were obtained by polymerising two different monomers in the appropriate molar ratio (see Table 1). Reaction conditions: (ⅰ) Ni(ClO4)2 (in EtOH), toluene, room temperature, 24–48 h, followed by precipitations in diisopropyl ether. | |
|
|
Table 1 Characterisation of the copolymers.a |
{kind=link}
{kind=link}
After purification, the molecular weights of the homopolymers and copolymers were determined by viscometry measurements. From the experimental intrinsic viscosities [η], Mv values were calculated using the phenomenological Mark-Houwink equation [η]= KMva. For constants K and a, we used K = 1.4 ×10-9 and a = 1.75, values that were previously determined for different polyisocyanides [23]. The results (Table 1) show that for all monomers and monomer combinations high molecular weight polymers were obtained. In this high molecular weight regime, the LCST and thus the gelation temperature does not depend on Mv any longer [17].
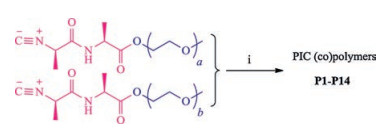
Rheology provided quantitative values of the LCSTs of the different polymer solutions/gels. To obtain values that are independent of frequency, we marked the onset temperature of the storage modulus increase as the gelation temperature (Tgel) (Fig. 2a). When plotted against the EG tail length, weight averaged by the fraction of each monomer, we find a clear linear correlation, at least for Tgel between 10 ℃ and 60 ℃ (Fig. 2b, dashed line, R2 = 98%). Such linear relation between the transition temperature and the tail length makes it straightforward to design hydrogels with customised gelation temperatures, for instance at body temperature (P11) or just below. Copolymer P2 (with 50% EG2 and 50% EG4) shows nearly the same transition temperature as homopolymer P3 with the same average length. Extrapolation of the observed linear relation reveals that the transition temperature of copolymer P1 (with 75% EG2 and 25% EG4) indeed is experimentally inaccessible, as it is close to 0 ℃. The graph also shows that small amounts of a monomer with relatively long tails (b = 8-12) has a large effect on the transition temperature.
The linear relation between Tgel and <nEG> was also observed in poly(meth)acrylates [15] and poly(vinyl ethers) that carry EG tails. In line with our results, for these polymers, the linearity does not hold anymore beyond 60 ℃. It is interesting to compare the relative contribution of a single EG group to the transition temperature, by looking at the slope of the curves in the linear regime. For the PIC polymers, the addition of a single EG group to the tail raises the transition temperature by circa 29 ℃. For methacrylate copolymers with long and short EG tails, lengthening the tail by one EG moiety increases the LCST with only 11 ℃. We speculate that the stronger effect of the PIC polymers is the result of the high grafting density; in PIC every backbone atom has an EG group attached, whereas in polymethacrylates and poly(vinyl ether)s, EG groups are grafted on every 2 or 3 backbone atoms. In addition, the helical conformation of the PIC backbone brings the tails close together, which may further strengthen the effect. Either way, the strong dependence requires careful tuning of the composition when a specific transition temperature is targeted. Fortunately, Fig. 2b shows that this is certainly possible, for instance, when aiming for a gelation temperature of 37 ℃.

|
Download:
|
| Fig. 2. Transition temperatures and mechanical properties of PIC (co)polymers and mixtures: (a) Rheology traces of selected polymers, colour coded for the monomers in the (co)polymer. (b) Transition temperatures of all polymers as a function of the average number of EG moieties in the tails. The colour coding is the same as in panel a. The dashed line is a linear fit through the experimental data with slope 28.9 ± 1.2. (c) Thermal and mechanical properties of the gelled mixture of P3 and P12 (purple data), compared to those of the individual gels (red and blue data, respectively). As a result of the continued stiffening of the gel with increasing temperature, a gel with lower Tgel (P3) will be stiffer than a gel with a higher Tgel (P12). The hybrid follows the same curve as P3. | |
{kind=link}
Solutions of polymers P2–P13 all form transparent hydrogels by heating through their respective LCSTs. Using oscillatory rheology, we determined the storage modulus G' (a measure for the elastic properties of the material) as well as the loss modulus G" (describing the viscous properties). At the LCST, G' shoots up a few orders of magnitude and in all cases, soft and elastic gels are formed (Fig. 2a) with G' >> G". The similarity between the polymers indicates that in all cases hydrogels of similar bundled architectures are formed, irrespective of the length of the EG tail. In other words, the mechanical properties are mostly determined by the bundled helical backbones. PIC gels, however, do continue to stiffen with increasing temperature, which means that at a set temperature, samples with lower gelation temperatures form stiffer hydrogels, see Fig. 2c with P3 (homopolymer with a = 3, Tgel = 18 ℃) and P12 (homopolymer with a = 4, Tgel = 42 ℃). For instance, at 50 ℃ a gel of P3 is nearly 100 times stiffer than a gel of P12 at the same concentration. Thus, tuning Tgel alloes one to readily tailor the mechanical properties. It should be noted that the same time, increase or reduce the gel stiffness [24].
Recently, we demonstrated that hybrid gels offer yet another layer of flexibility to tailor the mechanics of such (semi-flexible) materials [25-27]. We tested if gels with different transition temperatures would show hybrid behaviour, or whether they are incorporated into the same bundled network. To this end, we prepared a mixture of P3 and P12, co-dissolved them in cold water and followed gel formation of the mixture in a heating ramp (Fig. 2c, purple data). The resulting hybrid shows the thermal and mechanical properties of P3, which forms a gel at the lowest temperature of the two materials, but no significant change at the P12 transition temperature (42 ℃). The fact that the modulus of the hybrid closely follows that of P3 suggest that the P3 network is formed without interference of P12. As the stiffness of the P3 gel continues to increase with temperature, it will always be two orders of magnitude stiffer than the P12 gel, thereby dominating the mechanics of the hybrid. Microstructural analysis, vide infra, will show network formation of the second component.
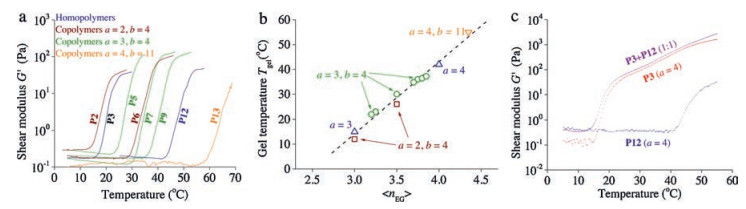
As the mechanical properties of the series follow the same trend, we argued that their microscopic architecture must be rather similar. Quantitative analysis of the architecture of PIC gels is difficult. Characteristic length scales in the gel (i.e., pore size and bundle diameters) are below the diffraction limit and AFM analysis or electron microscopy requires dehydration of the gel, which may strongly affect its structure. Cryo-based electron microscopy techniques allows us to visualise the network, but quantitative analysis of the images is challenging. Recently, we were able to insitu characterise the network structure using small angle X-ray scattering (SAXS) at the European Synchrotron Radiation Facilities in Grenoble. Appropriate models allowed us to retrieve hydrogel length scales from the scattering profiles of P3-based networks [28]. We now analysed the scattering patterns of the 1:1 mixture of P3 and P12 at three different temperatures (Fig. 3) by fitting them to the Kholodenko model for semi-flexible polymers and to the composite Kholodenko/Ornstein-Zernike model for semi-flexible bundles in a network structure (Supporting information). At the lowest temperature, where both polymers are homogeneously dissolved, the scattering profile shows the presence of individual semi-flexible chains in solution. The observed polymer chain radius (R = 0.9 nm) and persistence length (lp = 14.9 nm), obtained after fitting the data to the Kholodenko model for semi-flexible polymers, are in line with earlier obtained values measured for P3 [17]. The data at 35 ℃, i.e., above Tgel of P3 and below that of P12, requires fitting to a composite model of molecularly dissolved polymers (Kholodenko model for single polymers, with a fixed diameter R = 0.9 nm) and the network model, describing a porous structure with semi-flexible polymer bundles (Kholodenko/Ornstein-Zernike model) [28]. The fit yields two important parameters: Firstly, the average bundle size RB = 2.1 nm, which is slightly lower than the values that we find for gels of P3 without P12 present (RB = 3 nm) [17]; Secondly, the Ornstein-Zernike term gives the average length scale of network inhomogeneities, which approximates the pore size ξ. At 35 ℃, we find ξ = 51 nm, which compares well to earlier found values. At 50 ℃, we lose the molecularly dissolved polymer term in the scattering model and find a roughly unchanged average bundle size RB = 2.7 nm and, interestingly, a much smaller pore size ξ = 32 nm.

|
Download:
|
| Fig. 3. Small angle X-ray scattering pattern of a mixture of P3 and P12 at different temperatures: 5 ℃ (orange data), which is below Tgel of either polymer; at 35 ℃ (purple data) which is above Tgel of PIC3, but below that of PIC12 and at 50 ℃ (pink data), above Tgel of both compounds. The black lines are fits using the Kholodenko and the Ornstein-Zernike models (and their linear combinations, see text). The data at 35 ℃ and 50 ℃ was shifted vertically (10× and 100×, respectively) to increase clarity. | |
{kind=link}
The SAXS results indicate that indeed, P12 bundles beyond its gelation temperature and a hybrid is formed. Figs. 2a and c show that P3 and P12 display a similar jump in the mechanical properties at their respective transition temperatures. Due to the continued stiffening with temperature, however, at 45 ℃ (when the P12 gel is just formed), the P3 gel is already 100 times stiffer. Consequently, the mechanics of the weak gel simply remain hidden. After we strongly decreased the P3 concentration in the mixture to visualise the P12 contribution (ratio ~1:10), we observe hindered P3 gelation.
In conclusion, the thermal properties of thermoresponsive hydrogels in general and PIC gels in particular are a key parameter for their applications and often require fine-tuning. Usual approaches to tune the LCST include the addition of salts or other solutes and, of course, changing the polymer structure. For the class of EG-substituted polymers, the length of the EG tail strongly affects the LCST; in the case of PICs, it increases by nearly 30 ℃ for every additional ethylene glycol unit in the tail. This dependency is much stronger than for other EG-substituted thermoresponsive polymers, which means that to tailor the transition temperature, one can take advantages of the copolymerisation approach. With a transition temperature below 60 ℃, the LCST and also the gelation temperature of a random PIC copolymer is the average of the transition temperatures of the constituent homopolymers, weighed by the monomer fractions. This approach allows us to precisely tailor the gel mechanics at a desired temperature. A mixture (hybrid) of the two homopolymers, on the other hand, simply shows a transition for the component with the lowest Tgel. Beyond the gelation temperature of the second homopolymer, a hybrid network is formed, but when the mechanical properties of the two homopolymers are too far apart, the hybrid shows none of the potential mechanical advantages associated with these structures. In order to benefit from such features, one needs to tune the mechanical properties of the networks individually prior to mixing.
AcknowledgmentsWe acknowledge European Synchrotron Radiation Facility (ESRF) and their staff, in particular Dr. Daniel Hermida Merino, as well as the Netherlands Organisation for Scientific Research (NWO) for providing and supporting beam time at the DutchBelgium beamline (DUBBLE) for SAXS experiments (No. BM26-02773). We acknowledge financial support from NWO (VENI grant No. 680-47-437) and the Euopean Union's 2020 Research and Innovation Programme under Grant Agreement No. 642687, project Biogel.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2017.11.002.
| [1] |
D. Roy, W.L.A. Brooks, B.S. Sumerlin, Chem. Soc. Rev. 42(2013) 7214-7243. DOI:10.1039/c3cs35499g |
| [2] |
M.A. Ward, T.K. Georgiou, Polymers 3(2011) 1215. DOI:10.3390/polym3031215 |
| [3] |
N. Vanparijs, L. Nuhn, B.G. De Geest, Chem. Soc. Rev. 46(2017) 1193-1239. DOI:10.1039/C6CS00748A |
| [4] |
X. He, M. Aizenberg, O. Kuksenok, J. Aizenberg, et al., Nature 487(2012) 214-218. DOI:10.1038/nature11223 |
| [5] |
J.S. Scarpa, D.D. Mueller, I.M. Klotz, J. Am. Chem. Soc. 89(1967) 6024-6030. DOI:10.1021/ja01000a006 |
| [6] |
D.W. Urry, Sci. Am. 272(1995) 64-69. |
| [7] |
K. Kobayashi, C.I. Huang, T.P. Lodge, Macromolecules 32(1999) 7070-7077. DOI:10.1021/ma990242n |
| [8] |
G. Chen, A.S. Hoffman, Nature 373(1995) 49-52. DOI:10.1038/373049a0 |
| [9] |
S. Aoshima, H. Oda, E. Kobayashi, J. Polym. Sci. A:Polym. Chem. 30(1992) 2407-2413. DOI:10.1002/pola.1992.080301115 |
| [10] |
S. Han, M. Hagiwara, T. Ishizone, Macromolecules 36(2003) 8312-8319. DOI:10.1021/ma0347971 |
| [11] |
J.F. Lutz, J. Polym. Sci. A:Polym. Chem. 46(2008) 3459-3470. DOI:10.1002/(ISSN)1099-0518 |
| [12] |
H.R. Allcock, S.R. Pucher, M.L. Turner, R.J. Fitzpatrick, Macromolecules 25(1992) 5573-5577. DOI:10.1021/ma00047a002 |
| [13] |
T. Bauer, C. Slugovc, J. Polym. Sci. A:Polym. Chem. 48(2010) 2098-2108. DOI:10.1002/pola.v48:10 |
| [14] |
N. Sakai, M. Jin, S.I. Sato, T. Satoh, T. Kakuchi, Polym. Chem. 5(2014) 1057-1062. DOI:10.1039/C3PY00972F |
| [15] |
J.F. Lutz, A. Hoth, Macromolecules 39(2006) 893-896. DOI:10.1021/ma0517042 |
| [16] |
P.H.J. Kouwer, M. Koepf, V.A.A. Le Sage, et al., Nature 493(2013) 651-655. DOI:10.1038/nature11839 |
| [17] |
M. Jaspers, A.C.H. Pape, I.K. Voets, et al., Biomacromolecules 17(2016) 2642-2649. DOI:10.1021/acs.biomac.6b00703 |
| [18] |
M. Jaspers, M. Dennison, M.F.J. Mabesoone, et al., Nat. Commun 4(2014) 5808. |
| [19] |
M. Jaspers, P.H.J. Kouwer, Submitted(2017). |
| [20] |
R.K. Das, V. Gocheva, R. Hammink, O.F. Zouani, A.E. Rowan, Nat. Mater. 15(2016) 318-325. DOI:10.1038/nmat4483 |
| [21] |
M. Koepf, H.J. Kitto, E. Schwartz, et al., Eur. Polym. J. 49(2013) 1510-1522. DOI:10.1016/j.eurpolymj.2013.01.009 |
| [22] |
G. Hu, W. Li, Y. Hu, et al., Macromolecules 46(2013) 1124-1132. DOI:10.1021/ma302536t |
| [23] |
A. Van Beijnen, R. Nolte, W. Drenth, A. Hezemans, P. Van de Coolwijk, Macromolecules 13(1980) 1386-1391. DOI:10.1021/ma60078a011 |
| [24] |
M. Jaspers, A.E. Rowan, P.H.J. Kouwer, Adv. Funct. Mater. 25(2015) 6503-6510. DOI:10.1002/adfm.v25.41 |
| [25] |
M. Jaspers, S.L.P. Vaessen, P. van Schayik, et al., Nat. Commun. 8(2017) 15478. DOI:10.1038/ncomms15478 |
| [26] |
J.P. Gong, Soft Matter 6(2010) 2583-2590. DOI:10.1039/b924290b |
| [27] |
J.P. Gong, Y. Katsuyama, T. Kurokawa, Y. Osada, Adv. Mater. 15(2003) 1155-1158. DOI:10.1002/adma.200304907 |
| [28] |
D.C. Schoenmakers, A.E. Rowan, P.H.J. Kouwer, Submitted(2017). |