 2018, Vol. 29
2018, Vol. 29 



b Sino-Danish College, University of Chinese Academy of Science, Beijing 100049, China;
c Nano-Science Center and Department of Chemistry, University of Copenhagen, Copenhagen DK-2100, Denmark;
d Sino-Danish Center for Education and Research, Beijing 100049, China
Molecular electronics which illustrates the charge transport mechanism of individual or small groups of molecules provides fundamental investigation into the intrinsic molecular structureproperty relationship [1-4] and holds the most promise for the miniaturization of electronics [5, 6]. Recently, exciting research results such as the successful fabrication of unprecedented excellent single-molecule electrical switch [7], in-depth understanding of molecular interfacial spin effects along with the development and tailoring of molecular spinterface [8, 9], and the significant progresses in high-performance optoelectronics, especially in the field-effect transistors [10, 11], further stimulated the enthusiasm of researchers and good prospects for the future of molecular electronics. In order to efficiently bond to the two electrodes in the molecular sized nanogaps [12] and form the molecular junctions, anchoring groups are of unique importance in the fabrication and measurement of molecular electronics [13, 14]. A suitable anchoring group should have efficient binding and strong electronic coupling between molecule and electrode, so as to improve the charge transport, reduce the charge injection barrier across the metal-molecule interface and form reproducible and mechanically stable contacts. Sometimes, the anchoring group even showed a priority compared to the molecular backbone to determine hole-dominated (p-type) or electron-dominated (n-type) transport in the molecular junctions [15].
Thiol [13], amine [16], isocyanide [15], C60 [17] and many other groups [14] were widely used as the anchoring groups for bonding to different kinds of metal substrates to form high quality selfassembled monolayers (SAMs) and molecular electronics. Compared to thiol which was commonly used as anchoring group for molecular junctions, amine end groups were reported to be able to get more well-defined transport properties [18-20]. With a more restrict bond to Au substrate and electrode, amine groups can achieve better defined contact geometry in the molecular junctions. Thus, diamine molecules were used to comprehensively investigate the transport mechanism, as well as the statistical junction formation probability and stability [21-24]. With the advantage of forming amide bonds, amine anchors can also function as the efficient connectors for single molecular device based on the carbon electrodes such as graphene and carbon nanotubes [25-27]. More importantly, connecting single walled carbon nanotube (SWNT) or graphene as a springboard, amine anchors played an important role in exploring the electrical conductivity of individual DNA molecules [28, 29], the thermodynamic and kinetic behavior of a host-guest complex with high time resolution in a single molecule junction [30] and the further applications toward functional single-molecule photoswitches [31] and biochemical sensors [32].
Oligo(phenylene ethynylene)s (OPEs) with a highly conjugated and conductive backbone have been widely utilized in molecular electronics. Because of the ease of self-assembly, it is feasible to measure the conductance and charge transport mechanism in molecular junctions [21, 33]. And the conductance switching of OPE molecules may result from conformational changes [34-36]. In addition, an electron-rich and redox-active moiety of tetrathiafulvalene (TTF) group was also very attractive for switching the properties of molecular junctions [37, 38]. How about the combination of the OPEs and TTF? We have already developed the synthesis of the cruciform type system [39-42]. And OPE-TTFbased molecular junctions with dithiolate anchoring group measured by conducting probe-atomic force microscopy (CPAFM), scanning tunneling microscopy break-junction (STM-BJ) and mechanically controllable break-junction (MCBJ) were also reported recently [43-46]. The TTF substituent changes the molecular orbital energy levels, decreases the HOMO (highest occupied molecular orbital)-LUMO (lowest unoccupied molecular orbital) energy gap and achieves a better alignment of energy levels for charge injection, resulting in an increase of the conductance by an order of magnitude compared to the simple analogue OPE3 when studied by CP-AFM (monolayers), and, interestingly, these effects of TTF are also confirmed in the TTFpoly(p-phenylene ethynylene)s (PPEs) system [47]. However, we often found that the Au electrodes were strongly deformed such as the gold chains formed during the stretching process in the dithiolate anchoring conditions [48, 49]. Coupled with very flexible contact angle and chain/plane rotation, the variability of the observed conductance for the dithiol-Au junctions is relatively large. As mentioned above, amine group might be a favorable alternative to thiol because of its comparatively uniform contact geometry with both necessary angular flexibility for easy junction formation and a well-defined electronic coupling of the N lone pair to the Au [18, 50].
Herein, we used diamine as anchoring group for the OPE3 and OPE3-TTF (Fig. 1a) molecules to grow high quality SAMs and fabricate vertical molecular junctions by CP-AFM (Fig. 1b). Similar to the dithiolate anchored OPEs, TTF group also increased the molecular conductance of diamine OPE3 obviously. But the conductance difference induced by TTF substituent in diamine compounds was much larger than that of dithiolate. Besides the experimental results, such difference in electrical transport properties of the molecules was also analyzed theoretically.

|
Download:
|
| Fig. 1. (a) Molecular structures of OPE3 and OPE3-TTF. (b) Schematic of CP-AFM measurement of OPE3 SAMs. The molecules (OPE3 or OPE3-TTF) form SAMs on the Au substrate (short black wires represent Au–N bonds) through amine end groups (brick red circles). And fabricate vertical molecular junctions by CP-AFM for charge transport measurement. The purple part represents the AFM probe coated with metal (Au or Pt). | |
{kind=link}
Cyclic voltammetry (CV) measurement which is efficiently sensitive to the pinholes in the monolayer on metal surface [21, 51] was used to check the quality of SAMs. Fig. 2a showed the cyclic voltammograms (Fe2+/Fe3+ as redox active pair) of OPE3 and OPE3-TTF SAMs on Au. Compared to the bare Au electrode, the peaks for Fe2+/Fe3+ redox signal had almost disappeared for the SAMs covered Au surface. That implied both of the diamines formed highly dense SAMs which blocked the Fe2+/Fe3+ redox process. The current of OPE3-TTF SAM was slightly larger than the current observed for the OPE3 SAM, this result was due to relatively large TTF side chain and similar to their dithiolate analogues [43]. As the strength of the bond between anchor and substrate was also very important for the quality of SAMs, we also investigated the stability of SAMs by electrochemical reductive desorption as shown in Fig. 2b. Compared with the dithiolate molecules in control experiment, the diamine OPE3 and OPE3-TTF SAMs in this work had similar desorption values (Edesorption, ED) and sharp peaks refer to a double electron transfer process. That means the diamine anchoring group worked well in our OPE systems to form stable and uniform SAMs.

|
Download:
|
| Fig. 2. (a) Cyclic voltammograms and (b) electrochemical reductive desorption curves of OPE3 and OPE3-TTF SAMs on Au. | |
{kind=link}
The transport properties of the OPE3 and OPE3-TTF SAMs were measured by CP-AFM and compared. Both Au and Pt coated conductive tips were used as the top electrodes, thus symmetric Au/molecular wires/Au and asymmetric Pt/molecular wires/Au vertical device structures were formed. Fig. 3 showed the average Ⅰ–Ⅴ curves of about 200 molecular junction measurements of OPE3 and OPE3-TTF SAMs. We assumed that the contact areas in molecular junctions were almost the same in the whole set of measurements, as the same Au (or Pt) tip and soft contact load force value have been used in the all cases. Conductance histograms of the CP-AFM measured OPE3 and OPE3-TTF molecular junctions are shown in Figs. 3c and d. OPE3-TTF SAMs showed notably more than one order of magnitude higher current under both kinds of tips. We assign this effect to the TTF substituent which change the molecular orbital energy levels to decreases the HOMO-LUMO energy gap and contact resistance. The reduced HOMO-LUMO energy gap can be obtained from the UV–vis spectroscopy (Fig. S1 in Supporting information). Similar trend was found previously for the dithiolate OPE3 and OPE3-TTF, but the conductance difference resulting from the addition of the TTF moiety was much larger in this studied case having diamines anchoring group. The conductance values of the molecular junctions were determined over small voltage range (±0.1 V). The average conductance of OPE3-TTF SAM (168.87 nS) were 17 times higher than that of OPE3 SAM (9.65 nS) while using the Au tip. Under the asymmetric geometry with Pt tip, a 46 times difference was measured (the conductance was calculated to be 84.53 nS for OPE3-TTF SAM and 1.82 nS for OPE3 SAM). Higher conductance values came from the Au tip for both diamines, as the Au tip (which was made from commercial Pt coated AFM tip) had larger radius and thus more molecules could be caught in the devices. In addition, the conductance distribution of OPE3-TTF SAM was narrower than that of OPE SAM, which may be due to the incorporation of TTF increased the steric hindrance to reduce the torsion of the molecule, resulting in a more homogeneous SAM. Also, the conductance distribution of the diamine OPE3-TTF SAM was significantly narrower than that of dithiolate OPE3-TTF SAM [46], caused by the difference between the anchoring groups and demonstrating the excellent properties of the amine end capping groups.

|
Download:
|
| Fig. 3. Average Ⅰ–Ⅴ curve of about 200 molecular junctions based on the OPE SAMs measured by CP-AFM with (a) Au tip and (b) Pt tip. The insets showed the clear Ⅰ–Ⅴ curves of OPE3 with larger scale bar. Conductance histograms of the CP-AFM measured OPE3 molecular junctions with (c) Au tip and (d) Pt tip. | |
{kind=link}
As the previous study [43, 46, 52], the atomic structure of Au-SAM/probe electrode interface is ambiguous, and secondly in order to exclude any possible effects caused by the structure of the probe electrode, our theoretical model is based on the individual molecule chemisorbed between two Au electrodes.
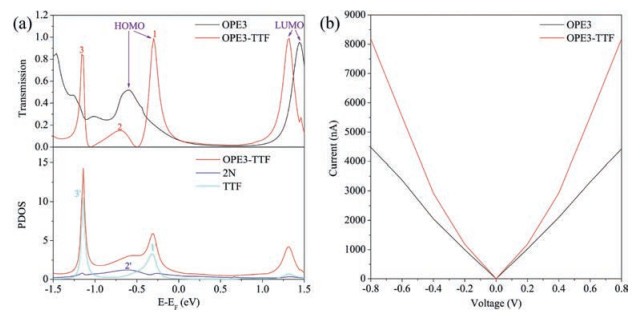
We calculated the transmission spectra of both the OPE3 and OPE3-TTF by using an equilibrium Green's function technique based on density functional theory (DFT), as implemented in the Atomistix ToolKit (ATK) package (see Supporting information) [53, 54]. The transmission spectrum (upper part of Fig. 4a) indicated that the energy of the LUMO is reduced and approached the Au Fermi level after the TTF group was introduced in OPE3 backbone. Furthermore, two new molecular orbitals are generated, corresponding to peaks 1 and 3, respectively. Wherein the molecular orbital corresponding to peak 1 became the HOMO of OPE3-TTF molecule, and because it is much closer to the Fermi level, the energy gap between HOMO and LUMO was decreased and the electron transport capacity of TTF-substituted molecules increased. We can explain this phenomenon through the projected density of states (PDOS) onto the nitrogen atoms and TTF (lower part of Fig. 4a). The PDOS onto the OPE3-TTF molecule was derived from the attachment of nitrogen atoms and TTF, and we found that it has a good agreement with the three transmission peaks 1, 2 and 3 in the transmission spectrum. The PDOS onto the nitrogen atoms corresponded to the transmission peak 2 (2'-2), and its energy position also coincided with the OPE3 device. The PDOS onto TTF corresponded to the transmission peaks 1 and 3 (1'-1, 3'-3), suggesting that the two transmission peaks were generated by TTF, so TTF can improve the electrical conductivity of device. In addition, the sharp transmission peaks 1 and 3 indicated that the coupling between the molecules and electrodes is relatively small, the increase in conductivity is mainly the contribution of TTF group. The result was also consistent with the molecular projected self-consistent Hamiltonian (MPSH) spectra (Fig. S2 in Supporting information), in which the incorporation of TTF remarkably reduced the coupling between the molecules and electrodes. Meanwhile, the higher electron density on the TTF substituent further confirmed that TTF dominated the electron transport.

|
Download:
|
| Fig. 4. (a) Calculated transmission through two OPEs (upper part) and PDOS onto the OPE3-TTF, 2N (nitrogen atoms of diamine) and TTF (lower part). (b) Calculated current through two OPEs. | |
{kind=link}
According to Landauer formula [55] (see Supporting information), we calculated the currents at different bias voltages. Through the intuitive Ⅰ–Ⅴ curve (Fig. 4b), we found that the TTF group can significantly increase the conductivity of the device, but the increased ratio is somewhat smaller than the experimental results. This may be due to differences in the nature of the electrodes in theoretical calculations and experiments, and/or many uncontrollable factors in the experiment, even the development of the theoretical simulations is not sufficient, as well as some of the complex and internal changes in the molecules or device that are not familiar to us formed during the formation of molecular junctions or the measurement process. However, we have achieved qualitative agreement between the theoretical and experimental value and this can explain the role of the TTF side group in the junction conductance.
Then we also calculated the transmission spectrum of dithiolate OPE3 as a comparison (Fig. S3a in Supporting information). The results showed that the HOMO and LUMO of the diamine OPE3 molecule move toward the high energy (right) direction relative to the dithiolate OPE3 molecule, resulting in the HOMO closer to the Au Fermi level, thereby increasing the electron transmission coefficient near the Fermi level. And the PDOS (Fig. S3b in Supporting information) onto the nitrogen atoms and sulfur atoms corresponded exactly to the transmission peaks. Besides, the Ⅰ–Ⅴ curves of diamine OPE3 and dithiolate OPE3 (Fig. S3c in Supporting information) also showed that the OPE3 molecules were connected to the Au through the nitrogen atoms had a stronger electron transport than the sulfur atoms connection. Also, the current values of the dithiolate OPE3 device at ±0.4 V were consistent with our previous theoretical calculation [46]. Meanwhile, these results also demonstrated that the larger conductance differences caused by the diamine groups that we have observed in Fig. 3, compared to the dithiolate anchoring groups.
In conclusion, high quality SAMs based on OPE3 and OPE3-TTF with diamine anchoring groups were grown on the Au substrate and fabricated into vertical molecular junctions by CP-AFM with two kinds of device geometries. The molecular conductance of diamine OPE3 was markedly increased by TTF group and the conductance differences were 17 and 46 times for Au and Pt tips, respectively, which were much higher than that of dithiolate anchored OPEs. Quantum chemical calculations qualitatively agreeing with the experimental data reproduced the substituent effect of TTF and the superiority of diamine anchoring groups. This system provides an opportunity to create well-defined and uniform SAMs, as well as to enhance the charge transport performance of molecular junctions. At the same time, the work has once again proved that the TTF substituents play a significant role in improving the conductance, and the switching caused by the insertion of TTF into OPEs has been reported in a threeterminal geometry [56]. OPE-TTFs thus show unique advantages of easily modified molecules as functional units in electronic circuits.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 61571415, 61622406, 51502283), the National Key Research and Development Program of China (Nos. 2017YFA0207500, 2016YFB0700700), and the "Hundred Talents Program" of Chinese Academy of Sciences (CAS).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.08.034.
| [1] |
Moth-Poulsen K., T. Bjørnholm, Nat. Nanotechnol. 4(2009) 551-556. DOI:10.1038/nnano.2009.176 |
| [2] |
H. Song, M.A. Reed, T. Lee, Adv. Mater. 23(2011) 1583-1608. DOI:10.1002/adma.201004291 |
| [3] |
T. Li, M. Jevric, J.R. Hauptmann, et al., Adv. Mater. 25(2013) 4164-4170. DOI:10.1002/adma.201300607 |
| [4] |
Z. Zhang, T. Li, Chin. Chem. Lett. 27(2016) 1209-1222. DOI:10.1016/j.cclet.2016.05.031 |
| [5] |
D. Xiang, X. Wang, C. Jia, T. Lee, X. Guo, Chem. Rev. 116(2016) 4318-4440. DOI:10.1021/acs.chemrev.5b00680 |
| [6] |
A. Cui, H. Dong, W. Hu, Small 11(2015) 6115-6141. DOI:10.1002/smll.v11.46 |
| [7] |
C. Jia, A. Migliore, N. Xin, et al., Science 352(2016) 1443-1445. DOI:10.1126/science.aaf6298 |
| [8] |
A. Cornia, P. Seneor, Nat. Mater. 16(2017) 505-506. DOI:10.1038/nmat4900 |
| [9] |
M. Cinchetti, V.A. Dediu, L.E. Hueso, Nat. Mater. 16(2017) 507-515. DOI:10.1038/nmat4902 |
| [10] |
J. Dong, H. Zhang, Chin. Chem. Lett. 27(2016) 1097-1104. DOI:10.1016/j.cclet.2016.05.005 |
| [11] |
Y. Zhen, H. Dong, L. Jiang, W. Hu, Chin. Chem. Lett. 27(2016) 1330-1338. DOI:10.1016/j.cclet.2016.06.023 |
| [12] |
T. Li, W. Hu, D. Zhu, Adv. Mater. 22(2010) 286-300. DOI:10.1002/adma.v22:2 |
| [13] |
J.C. Love, L.A. Estroff, J.K. Kriebel, R.G. Nuzzo, G.M. Whitesides, Chem. Rev. 105(2005) 1103-1170. DOI:10.1021/cr0300789 |
| [14] |
Moreno-García P., M. Gulcur, D.Z. Manrique, et al., J. Am. Chem. Soc. 135(2013) 12228-12240. DOI:10.1021/ja4015293 |
| [15] |
A. Tan, J. Balachandran, S. Sadat, et al., J. Am. Chem. Soc. 133(2011) 8838-8841. DOI:10.1021/ja202178k |
| [16] |
F. Chen, X. Li, J. Hihath, Z. Huang, N. Tao, J. Am. Chem. Soc. 128(2006) 15874-15881. DOI:10.1021/ja065864k |
| [17] |
C.A. Martin, D. Ding, J.K. Sørensen, et al., J. Am. Chem. Soc. 130(2008) 13198-13199. DOI:10.1021/ja804699a |
| [18] |
L. Venkataraman, J.E. Klare, I.W. Tam, et al., Nano Lett. 6(2006) 458-462. DOI:10.1021/nl052373+ |
| [19] |
I.S. Kristensen, D.J. Mowbray, K.S. Thygesen, K.W. Jacobsen, J. Phys.:Condens. Matter 20(2008) 374101. DOI:10.1088/0953-8984/20/37/374101 |
| [20] |
M.S. Hybertsen, L. Venkataraman, J.E. Klare, et al., J. Phys.:Condens. Matter 20(2008) 374115. DOI:10.1088/0953-8984/20/37/374115 |
| [21] |
Q. Lu, K. Liu, H. Zhang, et al., ACS Nano 3(2009) 3861-3868. DOI:10.1021/nn9012687 |
| [22] |
W. Hong, D.Z. Manrique, Moreno-García P., et al., J. Am. Chem. Soc. 134(2012) 2292-2304. DOI:10.1021/ja209844r |
| [23] |
M.T. González, A. Díaz, E. Leary, et al., J. Am. Chem. Soc. 135(2013) 5420-5426. DOI:10.1021/ja312392q |
| [24] |
M.T. González, X. Zhao, D.Z. Manrique, et al., J. Phys. Chem. C 118(2014) 21655-21662. DOI:10.1021/jp506078a |
| [25] |
C. Jia, J. Wang, C. Yao, et al., Angew. Chem. Int. Ed. 52(2013) 8666-8670. DOI:10.1002/anie.201304301 |
| [26] |
Y. Cao, S. Dong, S. Liu, et al., Angew. Chem. Int. Ed. 51(2012) 12228-12232. DOI:10.1002/anie.v51.49 |
| [27] |
X. Guo, Adv. Mater. 25(2013) 3397-3408. DOI:10.1002/adma.v25.25 |
| [28] |
X. Guo, A.A. Gorodetsky, J. Hone, J.K. Barton, C. Nuckolls, Nat. Nanotechnol. 3(2008) 163-167. DOI:10.1038/nnano.2008.4 |
| [29] |
S. Liu, G.H. Clever, Y. Takezawa, et al., Angew. Chem. Int. Ed. 50(2011) 8886-8890. DOI:10.1002/anie.v50.38 |
| [30] |
H. Wen, W. Li, J. Chen, et al., Sci. Adv. 2(2016) e1601113. DOI:10.1126/sciadv.1601113 |
| [31] |
C. Jia, J. Wang, C. Yao, Y. Cao, et al., Angew. Chem. Int. Ed. 52(2013) 8666-8670. DOI:10.1002/anie.201304301 |
| [32] |
L. Gao, L. L.-Li, X. Wang, et al., Chem. Sci. 6(2015) 2469-2473. DOI:10.1039/C4SC03612C |
| [33] |
L.A. Bumm, J.J. Arnold, M.T. Cygan, et al., Science 271(1996) 1705-1707. DOI:10.1126/science.271.5256.1705 |
| [34] |
J.L. Zhang, J.Q. Zhong, J.D. Lin, et al., Chem. Soc. Rev. 44(2015) 2998-3022. DOI:10.1039/C4CS00377B |
| [35] |
L.J. Wang, A. Yong, K.G. Zhou, et al., Chem.-Asian J. 8(2013) 1901-1909. DOI:10.1002/asia.v8.8 |
| [36] |
Z.J. Donhauser, B.A. Mantooth, K.F. Kelly, et al., Science 292(2001) 2303-2307. DOI:10.1126/science.1060294 |
| [37] |
S.H. Choi, C.D. Frisbie, J. Am. Chem. Soc. 132(2010) 16191-16201. DOI:10.1021/ja1060142 |
| [38] |
J. Liao, J.S. Agustsson, S. Wu, et al., Nano Lett. 10(2010) 759-764. DOI:10.1021/nl902000e |
| [39] |
M. Vestergaard, K. Jennum, J.K. Sørensen, K. Kilså, M.B. Nielsen, J. Org. Chem. 73(2008) 3175-3183. DOI:10.1021/jo702735d |
| [40] |
J.K. Sørensen, M. Vestergaard, A. Kadziola, K. Kilså, M.B. Nielsen, Org. Lett. 8(2006) 1173-1176. DOI:10.1021/ol060071o |
| [41] |
H. Lissau, R. Frisenda, S.T. Olsen, et al., Nat. Commun. 6(2015) 10233-10240. DOI:10.1038/ncomms10233 |
| [42] |
K. Jennum, M. Vestergaard, A.H. Pedersen, et al., Synthesis 4(2011) 539-548. |
| [43] |
Z. Wei, T. Li, K. Jennum, et al., Langmuir 28(2012) 4016-4023. DOI:10.1021/la204340n |
| [44] |
C.R. Parker, Z. Wei, C.R. Arroyo, et al., Adv. Mater. 25(2013) 405-409. DOI:10.1002/adma.201201583 |
| [45] |
C.R. Parker, E. Leary, R. Frisenda, etal., J.Am.Chem.Soc. 136(2014) 16497-16507. DOI:10.1021/ja509937k |
| [46] |
Z. Wei, T. Hansen, M. Santella, et al., Adv. Funct. Mater. 25(2015) 1700-1708. DOI:10.1002/adfm.201404388 |
| [47] |
Z. Wang, H. Dong, T. Li, et al., Nat. Commun. 6(2015) 8478-8487. DOI:10.1038/ncomms9478 |
| [48] |
Y. Kim, T.J. Hellmuth, M. Buerkle, F. Pauly, E. Scheer, ACS Nano 5(2011) 4104-4111. DOI:10.1021/nn200759s |
| [49] |
D. Kruger, R. Rousseau, H. Fuchs, D. Marx, Angew. Chem. Int. Ed. 42(2003) 2251-2253. DOI:10.1002/anie.200351000 |
| [50] |
S.Y. Quek, L. Venkataraman, H.J. Choi, et al., Nano Lett. 7(2007) 3477-3482. DOI:10.1021/nl072058i |
| [51] |
H. Valkenier, E.H. Huisman, van Hal P.A., et al., J. Am. Chem. Soc. 133(2011) 4930-4939. DOI:10.1021/ja110358t |
| [52] |
D. Fracasso, H. Valkenier, J.C. Hummelen, G.C. Solomon, R.C. Chiechi, J. Am. Chem. Soc. 133(2011) 9556-9563. DOI:10.1021/ja202471m |
| [53] |
J. Taylor, H. Guo, J. Wang, Phys. Rev. B 63(2001) 245407-245419. DOI:10.1103/PhysRevB.63.245407 |
| [54] |
M. Brandbyge, J.L. Mozos, P. Ordejon, J. Taylor, K. Stokbro, Phys. Rev. B 65(2002) 165401-165417. DOI:10.1103/PhysRevB.65.165401 |
| [55] |
M. Buttiker, Y. Imry, R. Landauer, S. Pinhas, Phys. Rev. B 31(1985) 6207-6215. DOI:10.1103/PhysRevB.31.6207 |
| [56] |
J. Fock, M. Leijnse, K. Jennum, et al., Phys. Rev. B 86(2012) 6244-6247. |