 2018, Vol. 29
2018, Vol. 29 



Noncovalent interactions are the key parts of supramolecu-larchemistry. The traditional noncovalent interactions such as hydrogen bond [1, 2], ionic interactions [3], van der Waals interactions [4], hydrophobic bonds [5-7] have been widely used to construct and operate functional systems. These weak interactions have been proved to play a fundamental role in some chemical and biological processes [8]. In the last two decades, the researchers have done a lot of efforts to explore new supramolecular weak interactions. The introduction of new noncovalent interactions is of significant importance in building functional systems and dictating the functionality of many biological systems [9]. Besides the traditional interactions, the ones involving aromatic rings have emerged enormously [8, 10, 11]. One typical example is π-π interactions which have been widely accepted and used in several supramolecular chemistry fields now. Cation-π interactions, as another example have attracted lots of interests since they were found around 20 years ago [10]. Many cation-π examples in nature have been found to support these interactions [11]. In biology and chemistry, cation-π interactions contribute significantly in many processes. For example, cation-π catalysis is found to be extremely important in the biosynthesis of steroids and organocatalysis [12-15]. This kind of interactions has played a central role in molecular recognition, translocation and transformation [10-18]. Compared with the cation-π interactions, anion-π interactions are much younger. As weak interactions at the forefront of interdisciplinary research, the discovery of anion-π interactions is different from the cation-π interactions. This noncovalent force between an electron-deficient π-system and an anionic moiety was firstly introduced by the theorists [19-21]. Then the experimental evidences in solid, solution and gas phase were gradually observed [9, 22-27]. The discussion about the nature of anion-π interactions is still undergoing together with an increasing amount of experimental investigations. The functional relevance study attracted much attention in the meantime. Transport of anion-π interactions appeared in functional systems around 10 years ago [28]. Now it is shifting to self-assembly on the one hand [29, 30] and catalysis on the other [31-39]. Like cation-π interactions, anion-π interactions are expected to become an essential part in chemistry and biochemical processes [26, 40-42]. In addition, their application in selective anion receptors, transport systems, self-assembly and catalysis definitively confirms their significance in the field of supramolecular chemistry. Other noncovalent interactions involving aromatic rings such as CH-π [43, 44] lone pair-π [45, 46] and salt-bridge-π are also at the forefront of research. In this review, we will only focus on the anion-π interactions. First of all, theoretical studies were introduced to give a point of view of the nature of anion-π interactions; and then, several recent and clear experimental evidence of anion-π interactions in solid, solution and gas phase was discussed; at last, some examples of applications especially in anion-π catalysis and self-assembly were described.
2. Theoretical studiesFrom the viewpoint of the theoretical study, the nature of anion-π is quite complicated. A general agreement is that anion-π interactions are mainly caused by electrostatic forces and ion-induced polarization [47-49]. It can be envisioned as anion-π interactions occur on the surface of electron-deficient aromatic planes with positive quadrupole moments. It is known that the aromatic rings with negative quadrupole moments are π-basic. Their electron-rich surfaces can attract cations, which are known as cation-π interactions [10-18].These interactions have beenwell recognized by now. On the other hand, to attract the anions, π-acidic aromatic rings are needed. The negative quadrupole moment has to be inverted to positive one. Normally this could be achieved by introducing electron-withdrawing substituents [9, 19-21, 25, 26, 30, 40, 50-57]. However, too much π-acidity will lead to charge-transfer complexes and radicals [58]. Deyà and co-workers studied the anion-π interactions by using a topological analysis of the electron density and MIPp calculations [20, 47]. They compared the typical example hexafluorobenzene with Qzz (quadrupole moment) = +9.5 B (B = Buckingham) and benzene with Qzz =-8.48B (Fig. 1). The introduction of the electron-withdrawing substituents-fluorine to the benzene ring inverts the quadrupole moment. The interactions of the electron-deficient aromatic rings and a series of anions were examined. A good correlation between the Qzz of the π-acidic rings and the electrostatic contribution to the total interaction energy was observed, which confirms that the electrostatic attractive interaction of anions with aromatic compounds is due to the crucial role of the Qzz. They also pointed out that the ion-induced polarization in anion-π interactions was also important.

|
Download:
|
| Fig. 1. Schematic representations of quadrupole moments of benzene and hexafluorobenzene. | |
{kind=link}
To acquire an efficient anion receptor, the aromatic ring should have a large positive quadrupole moment and large molecular polarizability. However, it is synthetically complicated to introduce more than three strong electron withdrawing groups and the spacer to the aromatic ring to build the receptor. The utilization of heteroaromatic rings could solve this problem. Mascal and coworkers studied the interactions between the electron-deficient heteroaromatic rings 1, 3, 5-triazine and trifluoro-1, 3, 5-triazine and fluoride, chloride, and azide ion by applying second-order Møller-Plesset perturbation theory (MP2) (Fig. 2) [19]. The electrostatic potential (ESP) maps were employed to visualize the charge distribution of aromatic rings under consideration (Fig. 2 down). They confirmed the favorable binding interactions between the N-heteroaromatic rings and the anions, with stronger interactions and shorter non-covalent bond distances.

|
Download:
|
| Fig. 2. 1, 3, 5-Triazine and trifluoro-1, 3, 5-triazine. Top: Schematic drawings; bottom: calculated ESP surfaces. Reprinted with permission [19]. Copyright 2002, American Chemical Society. | |
{kind=link}
3. Anion-π interactions in solid, solution and gas state
Since the first X-ray crystal structures with anion-π interactions were independently reported by Meyer et al. [59] and Reedijk et al. [60] in 2004, many examples especially metal complexes involving anion-π interactions between the counter anions and electron-deficient ligands in solid state have been reported [61-64]. Recently some new systems without metal ion have also been discovered [30, 65, 66].
Wang and co-workers developed a triazine based, electron-deficient and cavity self-tunable macrocyclic host—tetraoxacalix [2]arene[2]triazine (Fig. 3) [65]. It could form 1:1 complexes with four different kinds of anions (SCN-, NO3-, BF4-, PF6—), which were all confirmed by X-ray single-crystal diffraction. The formation of the anion-π complexes has also been evidenced in the gas phase through electrospray-ionization mass spectrometry (ESI-MS) measurements. In solution, fluorescence titration was done to give association constants for the formation of 1: 1 anion-π complexes in acetonitrile. This breakthrough indicates that this triazine based host could chelate anions with the two trizaine rings through the anion-π and lone-pair electron-π interactions.

|
Download:
|
| Fig. 3. Chemical structure of tetraoxacalix[2]arene[2]triazine and crystal structures of the anion-π complexes.Reprinted with permission [65]. Copyright 2013, American Chemical Society. | |
{kind=link}
Stoddart and co-workers reported a triangular prisms host based on naphthalene diimide(NDI) (Fig. 4a) [30]. The electron deficient cavities of the molecular prisms could be filled with linear I3- anions. The packing mode of the NDI-molecular prisms changed profoundly after introduction of I3- The inclusion of I3- anions induces π-π stacking of the chiral prisms into one handed supramolecular helices (Fig. 4b). This is a rare emergent example of anion-induced self-assembly with potential as ion-channels.

|
Download:
|
| Fig. 4. a) Structure of the rigid triangular molecular prism and single-crystal X-ray structure; b) Single-crystal X-ray structure of NDI host and [Bu4N][I3] complex. Reprinted with permission [30]. Copyright 2013, Wiley-VCH. | |
{kind=link}
The observations of the single-crystal complexes give a very strong support to the theoretical studies of anion-π interactions. However, the proximity in the solid state can originate from other effects besides anion-π interactions. Some more studies in the solution or gas state still need to be done. It is very difficult to observe and characterize the anion-π interactions in solution. The π-acidities of the receptors playakey role. Generally speaking, "too weak" anion-π interactions will not be observed. On the other hand, "too strong" one afford charge-transfer complexes and radicals [58]. The experimental evidence supporting the existence of attractive anion-π interactions involving charge-neutral aromatic systems in solution is still rare [25].
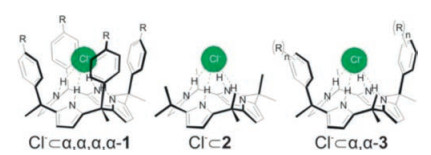
Ballester and co-workers have done great efforts to the experimental quantification of anion-π interactions in solution by using neutral host-guest model systems [25]. They developed several neutral molecular receptor systems based on a series of "two or four-wall" aryl-extended calix[4]pyrrole and a reference without wall (Fig. 5) [67, 68]. The NH groups of the calix[4]pyrrole could fix the halide anion in the aromatic cavity by hydrogen bond. Then the π-systems (wall) act as a receptor of anion-π interactions. By changing the π-acidity of the aromatic rings, a significant difference in free energy (ΔΔG) of binding between different complexes provides a direct measurement of the relative interaction energy of the halide with the different aromatic systems. This is a clear evidence for the existence ofboth repulsive and attractive interactions between π systems and halide anions in solution.

|
Download:
|
| Fig. 5. Molecular structures of the inclusion complexes of chloride with "four-wall" aryl extended calixpyrroles 1 (left), "no-wall" octamethylcalix[4]pyrrole 2 (middle), and "two-wall" aryl extended calix[4]pyrroles 3 (right). Reprinted with permission [68]. Copyright 2014, American Chemical Society. | |
{kind=link}
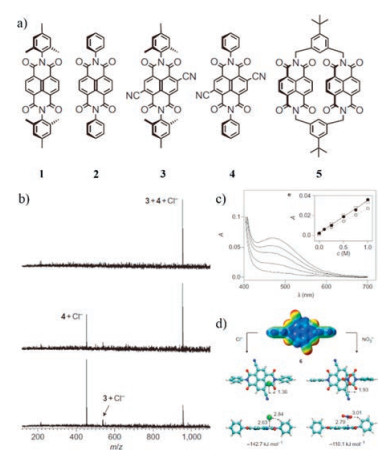
The first example of anion-π interactions in the gas phase was found by Hiraoka et al. in 1987 [69]. Stefan and co-worker reported another evidence in 2010 [50]. Naphthalenediimides (NDIs) with different π-acidity were chosen as anion receptors (Fig. 6a). Electrospray ionization fourier-transform ion cyclotron resonance tandem mass spectrometry (ESI-FTICR-MS-MS) was utilized to determine anion affinity and selectivity sequences quantitatively in combination with theoretical calculations (Figs. 6b and d). Competition experiments with NDI monomers 1-4 and chloride anion was done by the using mass spectrometry. The found selectivity sequence 4 > 3 > 2 > 1 demonstrated increasing anion affinity with increasing π-acidity and decrowding of the anion-π binding site. The ESI-FTICR-MS-MS technology can be used to determine anion affinity sequences quantitatively. These results provide a direct experimental evidence for anion-π interactions in the gas phase.

|
Download:
|
| Fig. 6. a) Structures of anion receptors naphthalene diimides (NDIs) 1–5; b) Laserinduced ESI-MS-MS fragmentation of heterodimer complexes; c) Charge-transfer absorption of the complexes; d) Molecular modelling of anion-π interactions. Reprinted with permission [50]. Copyright 2010, Nature Publishing Group. | |
{kind=link}
4. Anion-π interactions in functional systems
Anion-π interactions were applied to synthetic ion transporters in 2006 by Matile and co-workers [28]. This was the first example of anion-π interactions in functional systems. Now transport with anion-π interactions has been studied a lot. The attention is shifting to catalysis and self-assembly.
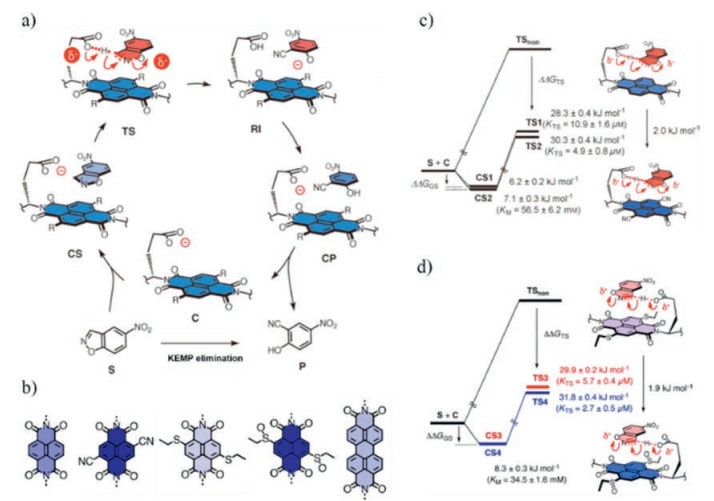
4.1. Anion-π catalysisIn 2013, Matile and co-workers reported the first example of anion-π catalysis [39]. Kemp elimination was selected as a sample model reaction which undergoes an anionic transition state (Fig. 7a). The anionic nature of the transition state could identify the contributions of the anion-π interactions. Naphthalene diimides (NDI) with different electron withdrawing groups like CN or SO and perylenediimide (PDI) acted as the electron-deficient surface. The stabilization of the anionic transition states increases with the π-acidity of the catalysts. This conclusion is verified by two separate series. The transition-state stabilization ΔΔGts for unsubstituted NDI and CN substituted NDI is 2.0kJ/mol (Fig. 7c) [39]. Similar result (1.9kJ/mol) is obtained from the sulfur or sulfoxide containing NDI catalysts (Fig. 7d) [38]. This study demonstrates that anion-π interactions indeed contribute to catalysis. It also broadens our understanding of organocatalysis and will lead to conceptually innovative design strategies to stabilize anionic transition states.

|
Download:
|
| Fig. 7. a) Catalytic cycle of the Kemp elimination with anion-π interactions; b) NDI or PDI based anion-π receptors; c, d) Energy diagram for the Kemp elimination catalyzed by different electron-deficient NDI. Reprinted with permission [38, 39]. Copyright 2014, American Chemical Society and 2013, Wiley-VCH. | |
{kind=link}
Matile and co-workers subsequently extended anion-π catalysis to enolate chemistry[35, 37, 70, 71]. They first gave an experimental evidence for the stabilization of reactive enolate intermediates by anion-π interactions. A malonate moiety was covalently attached on different π-acidic surface of NDIs (Fig. 8a) [37, 71]. The chemical shifts of the acidic hydrogen atoms of malonate on different π-acidic NDI surfaces were compared. The reactivity of these protons on π-acidic surfaces was measured by hydrogen-deuterium (H-D) exchange. The velocity of H-D exchange increases with π acidity. These results prove that enolates and their respective transition states could be stabilized by the π-acidic surfaces. Then they explored more significant enolate chemistry reactions. The addition ofMAHTs (1a) to enolate acceptors nitroolefin 2 fails to generate the additional product 3a (Fig. 8b). Instead, less useful thioester 4a as the major product is generated through favored decarboxylation. This enolate chemistry represents one of the most important anionic reactive intermediate in chemistry and biology. Here, different π-acidic NDIs surfaces were introduced for differentiating planar MHT tautomers with delocalized charges (as in reactive intermediate RI1, Fig. 8d) and deplanarizedtautomers with charges localized on the carboxylate (as in RI2) [70]. This discrimination influences the energy of the transition states leading to enolate addition and decarboxylation in different ways and thus to control the selectivity of the reaction. They also found that the position of the catalytic amine would influence the selectivity [35]. The rigidified Leonard turns is proved to be better than the flexible ones since it could address the MHT substrates close to the π-surface and make the reactions occur on aromatic surfaces and thus for harnessing the full potential of anion-π interaction for catalysis.

|
Download:
|
| Fig. 8. a) Positioning and stabilization of enolate anions on π-acidic surfaces; b) Addition of MAHT 1 to nitroolefin 2; c) The flexible turns place enolate tautomer far from the π surface; d) Transition state stabilization with the flexible turn catalyst; e) Fixed Leonard turns place the enolate tautomer close from the π surface; f) Transition state stabilization with the rigidified Leonard turn catalyst. Reprinted with permission [35, 37]. Copyright 2014, Nature Publishing Group and 2016, Wiley-VCH. | |
{kind=link}
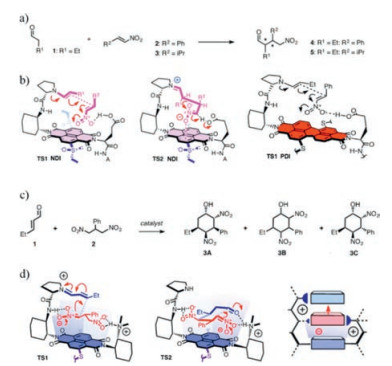
Matile and co-workers have taken one step further the research on anion-π catalysis by extending it to enamine and iminium chemistry [33, 36, 72]. A proline forenamine formation is located on one side and a glutamic acid for nitronate protonation is on the other side of the π-surface. The enamine addition to nitroolefins occurs on the aromatic surface of π-acidic naphthalene diimides or perylenediimide (Fig. 9a and b) [36, 72]. With the increase of π acidity of the NDI or PDI catalysts, rate and enantioselectivity of the reaction increases. This is the first example of anion-π asymmetric catalysis. The chiral sulfoxides substituent at the edge of the π-acidic surface has a profound influence on asymmetric anion-π catalysis. A perfect matched structure could afford the highest enantio-and diastereo-selectivity. For iminium chemistry catalysis, a more demanding cascade process is achieved byasymmetric anion-π catalysis. The selected example affords six-membered carbocycles with five stereogenic centers in a single step from achiral and acyclic substrates (Fig. 9c) [33]. Similar to anion-π catalysis in enamine chemistry, rates and stereoselectivity increase with the π-acidity. This gave another experimental support for the contribution of anion-π interactions to the stereoselective stabilization of the anionic transition states.

|
Download:
|
| Fig. 9. a) Selected enamine chemistry of aldehyde to nitroolefins; b) Possible stabilization of adding enamines to nitroolefins (TS1) and nitronate protonation (TS2) on the π-acidic surface (NDI or PDI) of anion-π catalyst; c) Cascade reaction catalyzed by anion-π systems; d) Transition states stabilization (TS1, TS2 and schematic side view). Reprinted with permission [33, 36, 72]. Copyright 2015, 2016, American Chemical Society and 2016, Wiley-VCH. | |
{kind=link}
Since the report of anion-π catalysis for the first time in 2013, it has been explored with enolate, enamine, iminium and so on. Very recently, the first anion-π enzyme has been created [34]. The idea of stabilizing anionic intermediates and transition states on π-acidic surfaces has become a new fundamental concept. More and more neworganic reactions will be successfully catalyzed with anion-π interactions. New electron deficient p surface needs to be explored to extend the anion-π catalysts with other complex systems. High expectations exist in the catalysis of some important reactions both in chemistry and biology which cannot be realized without anion-π or other weak interactions.
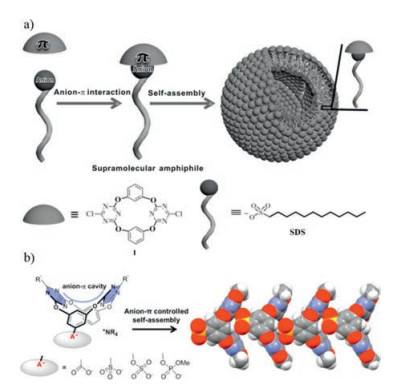
4.2. Self-assembly with anion-π interactionsWang and co-workers reported an example of self-assembly induced by anion-π interactions [65, 73]. The neutral tetraoxacalix [2]arene[2]triazine has been studied very well as an anion-π receptor in solution or solid state [74-76]. Here it acted as a host and hydrophobic anions such as sodium dodecyl sulfate (SDS), laurate, and so on as guests [29]. Supramolecular amphiphiles were found to self-assemble into vesicles in water through anion-π interactions. Decreasing the π-acidity of the host by introducing donating group amines instead of chloride led to neither micelles nor vesicles. The disassembly of the vesicles was promoted by the addition of competing anions such as NO3-, Cl- and Br- or a decrease in the pH value. The same group recently reported another anion-π induced self-assembly example [73]. An anionic head A- (Fig. 10) such as carboxylate, sulfonate, sulfate, and phosphate was introduced to the host skeleton. The triazine rings provide an electron-deficient cavity. The formation of head-to-tail oligomeric aggregation both in solution and solid state was obtained. Anion-π interaction between the anionic head and the electron-deficient cavity plays a decisive role to direct the assembly formation.

|
Download:
|
| Fig. 10. a) Schematic diagram of the formation of vesicles as induced by anion-π interactions; b) Incorporating an anionic head onto oxacalix[2]arene[2]-triazine for anion-π directed assembly. Reprinted with permission [29, 73]. Copyright 2015, Wiley-VCH and 2017, American Chemical Society. | |
{kind=link}
5. Summary and outlook
Anion-π interactions as a new member of supramolecular weak interactions have attracted considerable attentions. From the beginning the theorists put forward the concept, experimental evidence was gradually found in solid, solution and gas state. Numerous studies focus on the host-guest recognition. There are sufficient experimental evidence now to validate this new interaction. The research went through theoretical investigations; recognition in solid, solution or gas state; functionalization and so on. Recalling the development of cation-π interactions, it is only 20 years since it was found. Now cation-π interactions play a salient role in supramolecular chemistry and have been widely used in recognition and organocatalysis. In analogy to cation-π interactions, the study about anion-π interactions is also shifting to the construction of functional systems. Especially the current emergence of anion-π catalysis brings exciting opportunities. More and more functional systems operating with anion-π interactions will be realized. The objective of this mini-review is to highlight the recent developments in anion-πrecognition and functionalization, emphasizing the great functionalized potential of anion-π interactions particularly in catalysis.
AcknowledgmentsThe work was supported by the National Natural Science Foundation of China (No. 21604046), the National Young Thousand Talents Program, Shandong Provincial Natural Science Foundation, China (No. ZR2016XJ004).
| [1] |
E. Arunan, G.R. Desiraju, R.A. Klein, et al., Pure Appl. Chem. 83(2011) 1619-1636. |
| [2] |
M.C. Qi Wang, J.L. Jiang, L.Y. Wang, Chin. Chem. Lett. 28(2017) 793-797. DOI:10.1016/j.cclet.2017.02.008 |
| [3] | |
| [4] | |
| [5] |
D. Chandler, Nature 437(2005) 640-647. |
| [6] |
X.F. Ji, P. Wang, H. Wang, F.H. Huang, Chin. J. Pol. Sci. 33(2015) 890-898. DOI:10.1007/s10118-015-1639-6 |
| [7] |
Z.J. Yin, Z.Q. Wu, F. Lin, et al., Chin. Chem. Lett. 28(2017) 1167-1171. |
| [8] |
E.A. Meyer, R.K. Castellano, F. Diederich, Angew. Chem. Int. Ed. 42(2003) 1210-1250. |
| [9] |
L.M. Salonen, M. Ellermann, F. Diederich, Angew. Chem. Int. Ed. 50(2011) 4808-4842. DOI:10.1002/anie.v50.21 |
| [10] |
D.A. Dougherty, Science 271(1996) 163-168. |
| [11] |
D.A. Dougherty, Acc. Chem. Res. 46(2013) 885-893. DOI:10.1021/ar300265y |
| [12] |
Q. Zhang, K. Tiefenbacher, Nat. Chem. 7(2015) 197-202. |
| [13] |
M.C. Holland, J.B. Metternich, C. Mueck-Lichtenfeld, R. Gilmour, Chem. Commun. 51(2015) 5322-5325. |
| [14] |
R.R. Knowles, S. Lin, E.N. Jacobsen, J. Am. Chem. Soc. 132(2010) 5030-5032. DOI:10.1021/ja101256v |
| [15] |
S. Yamada, J.S. Fossey, Org. Biomol. Chem. 9(2011) 7275-7281. DOI:10.1039/c1ob05228d |
| [16] |
J.A. Faraldos, A.K. Antonczak, V. Gonzalez, et al., J. Am. Chem. Soc. 133(2011) 13906-13909. |
| [17] |
D.A. Stauffer, R.E. Barrans, D.A. Dougherty, Angew. Chem. Int. Ed. 29(1990) 915-918. DOI:10.1002/(ISSN)1521-3773 |
| [18] |
K.U. Wendt, G.E. Schulz, E.J. Corey, D.R. Liu, Angew. Chem. Int. Ed. 39(2000) 2812-2833. |
| [19] |
M. Mascal, A. Armstrong, M.D. Bartberger, J. Am. Chem. Soc. 124(2002) 6274-6276. DOI:10.1021/ja017449s |
| [20] |
D. Quinonero, C. Garau, C. Rotger, et al., Angew. Chem. Int. Ed. 41(2002) 3389-3392. DOI:10.1002/1521-3773(20020916)41:18<3389::AID-ANIE3389>3.0.CO;2-S |
| [21] |
I. Alkorta, I. Rozas, J. Elguero, J. Am. Chem. Soc. 124(2002) 8593-8598. DOI:10.1021/ja025693t |
| [22] |
D. Quinonero, A. Frontera, D. Escudero, et al., ChemPhysChem 8(2007) 1182-1187. DOI:10.1002/(ISSN)1439-7641 |
| [23] |
M. Giese, M. Albrecht, K. Rissanen, Chem. Rev. 115(2015) 8867-8895. DOI:10.1021/acs.chemrev.5b00156 |
| [24] |
H.T. Chifotides, K.R. Dunbar, Acc. Chem. Res. 46(2013) 894-906. DOI:10.1021/ar300251k |
| [25] |
P. Ballester, Acc. Chem. Res. 46(2013) 874-884. |
| [26] |
A. Frontera, P. Gamez, M. Mascal, T.J. Mooibroek, J. Reedijk, Angew. Chem. Int. Ed. 50(2011) 9564-9583. |
| [27] |
M. Giese, M. Albrecht, K. Rissanen, Chem. Commun. 52(2016) 1778-1795. DOI:10.1039/C5CC09072E |
| [28] |
V. Gorteau, G. Bollot, J. Mareda, A. Perez-Velasco, S. Matile, J. Am. Chem. Soc. 128(2006) 14788-14789. DOI:10.1021/ja0665747 |
| [29] |
Q. He, Y.F. Ao, Z.T. Huang, D.X. Wang, Angew. Chem. Int. Ed. 54(2015) 11785-11790. DOI:10.1002/anie.201504710 |
| [30] |
S.T. Schneebeli, M. Frasconi, Z. Liu, et al., Angew. Chem. Int. Ed. 52(2013) 13100-13104. DOI:10.1002/anie.201307984 |
| [31] |
L. Liu, Y. Cotelle, J. Klehr, et al., Chem. Sci. 8(2017) 3770-3774. DOI:10.1039/C7SC00525C |
| [32] |
Y. Zhao, Y. Cotelle, N. Sakai, S. Matile, J. Am. Chem. Soc. 138(2016) 4270-4277. DOI:10.1021/jacs.5b13006 |
| [33] |
L. Liu, Y. Cotelle, A.-J. Avestro, N. Sakai, S. Matile, J. Am. Chem. Soc. 138(2016) 7876-7879. DOI:10.1021/jacs.6b04936 |
| [34] |
Y. Cotelle, V. Lebrun, N. Sakai, T.R. Ward, S. Matile, ACS Cent. Sci. 2(2016) 388-393. |
| [35] |
Y. Cotelle, S. Benz, A.-J. Avestro, et al., Angew. Chem. Int. Ed. 55(2016) 4275-4279. |
| [36] |
Y. Zhao, Y. Cotelle, J. A.-Avestro, N. Sakai, S. Matile, J. Am. Chem. Soc. 137(2015) 11582-11585. DOI:10.1021/jacs.5b07382 |
| [37] |
Y. Zhao, N. Sakai, S. Matile, Nat. Commun 5(2014). |
| [38] |
Y. Zhao, C. Beuchat, Y. Domoto, et al., J. Am. Chem. Soc. 136(2014) 2101-2111. |
| [39] |
Y. Zhao, Y. Domoto, E. Orentas, et al., Angew. Chem. Int. Ed. 52(2013) 9940-9943. DOI:10.1002/anie.201305356 |
| [40] |
C. Estarellas, A. Frontera, D. Quinonero, P.M. Deya, Angew. Chem. Int. Ed. 50(2011) 415-418. DOI:10.1002/anie.201005635 |
| [41] |
S. Chakravarty, Z.Z. Sheng, B. Iverson, B. Moore, FEBS Lett. 586(2012) 4180-4185. DOI:10.1016/j.febslet.2012.10.017 |
| [42] |
D.D. Jenkins, J.B. Harris, E.E. Howell, R.J. Hinde, J. Baudry, J. Comput. Chem. 34(2013) 518-522. DOI:10.1002/jcc.23164 |
| [43] | |
| [44] |
M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama, H. Suezawa, CrystEngComm 11(2009) 1757-1788. DOI:10.1039/b902318f |
| [45] |
M. Egli, S. Sarkhel, Acc. Chem. Res. 40(2007) 197-205. DOI:10.1021/ar068174u |
| [46] |
T.J. Mooibroek, P. Gamez, J. Reedijk, CrystEngComm 10(2008) 1501-1515. DOI:10.1039/b812026a |
| [47] |
C. Garau, A. Frontera, D. Quinonero, et al., ChemPhysChem 4(2003) 1344-1348. DOI:10.1002/cphc.v4:12 |
| [48] |
D. Quiñonero, C. Garau, A. Frontera, et al., Chem. Phys. Lett. 359(2002) 486-492. DOI:10.1016/S0009-2614(02)00709-1 |
| [49] |
A. Bauza, P.M. Deya, A. Frontera, D. Quinonero, Phys. Chem. Chem. Phys. 16(2014) 1322-1326. DOI:10.1039/C3CP54147A |
| [50] |
R.E. Dawson, A. Hennig, D.P. Weimann, et al., Nat. Chem. 2(2010) 533-538. DOI:10.1038/nchem.657 |
| [51] |
L. Adriaenssens, C. Estarellas, A.V. Jentzsch, et al., J. Am. Chem. Soc. 135(2013) 8324-8330. DOI:10.1021/ja4021793 |
| [52] |
B.L. Schottel, H.T. Chifotides, K.R. Dunbar, Chem. Soc. Rev. 37(2008) 68-83. |
| [53] |
H.T. Chifotides, B.L. Schottel, K.R. Dunbar, Angew. Chem. Int. Ed. 49(2010) 7202-7207. DOI:10.1002/anie.v49:40 |
| [54] |
M.M. Watt, M.S. Collins, D.W. Johnson, Acc. Chem. Res. 46(2013) 955-966. DOI:10.1021/ar300100g |
| [55] |
A.V. Jentzsch, A. Hennig, J. Mareda, S. Matile, Acc. Chem. Res. 46(2013) 2791-2800. |
| [56] |
M.M. Watt, L.N. Zakharov, M.M. Haley, D.W. Johnson, Angew. Chem. Int. Ed. 52(2013) 10275-10280. DOI:10.1002/anie.201303881 |
| [57] |
M. Giese, M. Albrecht, K. Wiemer, et al., Eur. J. Inorg. Chem(2012), 2995-2999. |
| [58] |
S. Guha, F.S. Goodson, L.J. Corson, S. Saha, J. Am. Chem. Soc. 134(2012) 13679-13691. DOI:10.1021/ja303173n |
| [59] |
S. Demeshko, S. Dechert, F. Meyer, J. Am. Chem. Soc. 126(2004) 4508-4509. DOI:10.1021/ja049458h |
| [60] |
P. de Hoog, P. Gamez, H. Mutikainen, U. Turpeinen, J. Reedijk, Angew. Chem. Int. Ed. 43(2004) 5815-5817. DOI:10.1002/(ISSN)1521-3773 |
| [61] |
H.T. Chifotides, I.D. Giles, K.R. Dunbar, J. Am. Chem. Soc. 135(2013) 3039-3055. |
| [62] |
C.S. Campos-Fernandez, B.L. Schottel, H.T. Chifotides, et al., J. Am. Chem. Soc. 127(2005) 12909-12923. DOI:10.1021/ja052108q |
| [63] |
B.L. Schottel, H.T. Chifotides, M. Shatruk, et al., J. Am. Chem. Soc. 128(2006) 5895-5912. DOI:10.1021/ja0606273 |
| [64] |
C.A. Black, L.R. Hanton, M.D Spicer, Chem. Commun.(2007), 3171-3173. |
| [65] |
D.X. Wang, M.X. Wang, J. Am. Chem. Soc. 135(2013) 892-897. DOI:10.1021/ja310834w |
| [66] |
N. Hafezi, J.M. Holcroft, K.J. Hartlieb, et al., Angew. Chem. Int. Ed. 54(2015) 456-461. |
| [67] |
G. Gil-Ramirez, E.C. Escudero-Adan, J. Benet-Buchholz, P. Ballester, Angew. Chem. Int. Ed. 47(2008) 4114-4118. DOI:10.1002/(ISSN)1521-3773 |
| [68] |
L. Adriaenssens, G. Gil-Ramirez, A. Frontera, et al., J. Am. Chem. Soc. 136(2014) 3208-3218. DOI:10.1021/ja412098v |
| [69] |
K. Hiraoka, S. Mizuse, S. Yamabe, J. Phys. Chem. 91(1987) 5294-5297. DOI:10.1021/j100304a032 |
| [70] |
Y.J. Zhao, S. Benz, N. Sakai, S. Matile, Chem. Sci. 6(2015) 6219-6223. DOI:10.1039/C5SC02563J |
| [71] |
F.N. Miros, Y. Zhao, G. Sargsyan, et al., Chem.-Eur. J. 22(2016) 2648-2657. DOI:10.1002/chem.201504008 |
| [72] |
C. Wang, F.N. Miros, J. Mareda, N. Sakai, S. Matile, Angew. Chem. Int. Ed. 55(2016) 14420-14424. |
| [73] |
R.B. Xu, Q.Q. Wang, Y.F. Ao, et al., Org. Lett. 19(2017) 738-741. DOI:10.1021/acs.orglett.7b00070 |
| [74] |
D.X. Wang, Q.Y. Zheng, Q.Q. Wang, M.X. Wang, Angew. Chem. Int. Ed. 47(2008) 7485-7488. |
| [75] |
D.X. Wang, Q.Q. Wang, Y. Han, et al., Chem.-Eur. J. 16(2010) 13053-13057. DOI:10.1002/chem.201002307 |
| [76] |
D.X. Wang, S.X. Fa, Y. Liu, B.Y. Hou, M.X. Wang, Chem. Commun. 48(2012) 11458-11460. DOI:10.1039/c2cc36465d |