 2018, Vol. 29
2018, Vol. 29 



Methane, with abundant reserves on earth, is regarded as the most promising clean fossil energy [1]. In conventional methane combustion processes, the flame temperature can reach up to 1100 ℃, which inevitably leads to additional energy waste and produces pollutant gases (such as nitrogen oxides (NOx), carbon monoxide (CO)) [2]. Recently, the boost of natural gas vehicles also promotes the increased utilization of earth-abundant natural gas energy [3], which leads to a large amount of methane emissions and thus aggravates its environmental burden. Methane emissions from automobile exhaust and coal mine have also contributed to the greenhouse effect and global warming [3, 4]. Due to the increasing energy efficiency requirements and stringent environmental emission regulations, improving high utilization efficiency of methane is an urgent issue needed to be addressed.
Heterogeneous catalytic combustion of methane has been reported to be able to improve the energy utilization efficiency of methane and control unnecessary atmospheric pollution [2, 5, 6]. Supported palladium oxides exhibit outstanding catalytic performance for methane combustion [7]. However, metal oxide supported PdO catalysts tend to deactivate below 450 ℃, which seriously hinders their long-term application in catalytic combustion. It is likely that accumulation of hydroxyls on the supports prevent the migration of oxygen to active Pd sites and thus inhibits the catalytic reaction [8]. Moreover, the high cost of Pd also limits its broad application in methane catalytic combustion and methane emission control [9, 10].
Recently, accompanied with the development of nanomaterials synthesis technology, nanostructured perovskite ABO3-type materials with increased surface area have received renewed attention as a potential industrial catalyst for exhaust emission control [11]. They were proposed as potential non-precious metal catalysts in methane catalytic combustion due to excellent hydrothermal stability and sulfur resistance compared with Pd-based catalysts. Manganese-and cobalt-based perovskites have been proven to show excellent catalytic activity for oxidation (combustion) reactions [12-14]. The concentration of adsorbed oxygen, the activity of lattice oxygen and redox properties of B-site ions are closely related to the catalytic properties of perovskite oxides. In this review, we have summarized the recent research progress on nanostructured perovskite oxides for catalytic methane combustion as well as the existing issues need to be addressed in the near future.
2. Mechanism for methane combustionSignificant efforts have been devoted to elucidating the mechanism of methane combustion on different types of catalysts. Breakage of the first C-H bond is regarded as the first and the ratedependent step for methane catalytic combustion due to its extremely high activation energy (439.3 eV). Noble metals and transition-metal oxides based catalysts have been widely studied for catalytic combustion of methane. Pd based catalysts have been well-studied as high-performance catalysts for CH4 combustion. Chin and Iglesia et al. [15] expounded an explicit mechanistic pathway for C-H bond activation from the perspective of theory and experiment. The catalytic reactivity for C-H bond activation varied with Pd chemical states, O centers involved and surface oxygen coverages. Chemical potential of oxygen was closely related to the extent of C-H bond activation. The higher oxygen chemical potential, the easier C-H bond activation on the catalysts surface.
In addition to noble metal based catalysts, single-metal-oxidebased catalysts (such as copper oxide (CuO) [16] along with MnOxbased catalysts [17] and spinel [3]) were also investigated as efficient catalysts for methane combustion. Feng et al. [16] have first reported the study of the catalytic combustion of methane over CuO nanowires (NWs). The conversion rate of CH4 over the catalytic CuO NWs was almost 40% at 500 ℃ and the catalytic activity of the CuO NWs could remain stable for at least 24 h at 500 ℃, which proved that CuO NWs have great potential to be effective catalysts for methane combustion. Zhang et al. [17] prepared MnOx-NiO composite oxide catalysts via co-precipitation method, which exhibited superior catalytic performance in the lean methane oxidation due to good reducibility and oxygen mobility. Tao et al. [3] had observed that NiCo2O4 exhibited better catalytic activity than precious metal-catalysts. The CH4 activation mechanism was also different: CH4 first dissociated to –CH3 on metal sites (Ni3+), then coupled with a surface lattice oxygen (O2-) to form –CH3O; and it dehydrogenized to –CHO for successive two steps; finally –CHO was converted into CO2.
Compared with the well-studied CH4 catalytic oxidation mechanism on NiCo2O4, mechanistic understanding of the methane combustion over perovskite-type catalysts is still under debating. Pena and Fierro et al. [18] firstly investigated the kinetics of methane catalytic oxidation over perovskites catalysts. Eley Rideal (E-R) mechanism, in which gaseous methane reacted with dissociative adsorbed oxygen species on the perovskite surface, was proposed. Then, Auer and Thyrion et al. [19] had conducted another detailed kinetic analysis: (1) CH4 first adsorbed on the catalyst surface to form adsorbed CH4(ad); (2) Dioxygen fast transformed into active oxygen species O* and O* reacted with CH4(ad) to form intermediate Is; (3) Finally Is quickly transformed into adsorbed CO2(ad) and H2O(ad), and then CO2(ad) and H2O(ad) desorbed to molecular CO2 and H2O. The oxidation rate of methane was controlled by desorption of products molecules (CO2 and H2O). According to their calculation, the Mars and van Krevelen mechanism (MvK mechanism) was more suitable pathway for methane combustion. The methane oxidation process was mainly dependent on lattice oxygen (O2-) in perovskite oxides [20]. Fig. 1 illustrated the methane oxidation process through MvK pathway: (1) The methane in the gas phase first adsorbed on the A site to form CH4(ad); (2) CH4(ad) was attacked by surface lattice oxygen to form CH3 and the latter was continuously oxidized by surface lattice oxygen to generate CO2(ad) and H2O(ad) with the generation of oxygen vacancies; (3) CO2(ad) and H2O(ad) desorbed from the catalysts surface to form molecule CO2 and H2O, and lattice oxygen was regenerated by refilling of oxygen vacancies with surface adsorbed oxygen. In addition, the amount of active oxygen species also plays an important role in methane catalytic combustion. However, further quantitative investigation on each step of MvK mechanism is needed to obtain a perspicuous understanding.

|
Download:
|
| Fig. 1. The pathways of CH4 combustion on ABO3-type perovskites following MvK mechanism. | |
3. Rational design of perovskite oxide catalysts
The typical ABO3-type perovskite oxides show an ideal cubic structure unit cell with space group Pm3m-Oh in Fig. 2a [21], in which the larger A ion is in dodecahedral coordination with oxygen and the smaller B ion is 6-fold coordinated toward oxygen ion. Due to its stable crystal structure, the A and B cations in perovskites could be partially substituted and give rise to substituted compounds with formula of A1-xA'xB1-xB'xO3 [18]. As shown in Fig. 2b [22], A-or B-site substitution in perovskites has also brought the change of metal ions' oxidation states in the crystal structure and led to the generation of oxygen vacancies. The vast majority of metallic elements from the periodic table could be used as substituted ions, which further gives rise to the great flexibility of perovskites constitutions. The broad family of perovskites oxides also provides the potential for their application in various catalytic processes. Furthermore, the unique structure of perovskite oxides has the possibility to incorporate other types of A or B ions with suitable size and charge, which could change the valence state and nature of lattice ions, thus leading to the tailorable redox and physicochemical properties.

|
Download:
|
| Fig. 2. (a) Schematic illustration of an ideal ABO3 perovskite unit cell. Copied with permission [21]. Copyright 2015, American Chemical Society. (b) Crystal model of A site doped ABO3 perovskite with oxygen vacancy. Copied with permission [22]. Copyright 2009, Elsevier B.V. | |
3.1. Design of redox properties
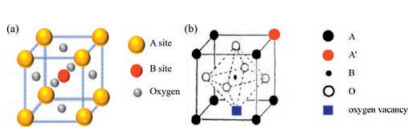
The catalytic activity of perovskite oxides is closely related to their redox properties [23, 24]. And the intrinsic redox properties of perovskite oxides are generally determined by B-site cations properties. Temperature-programmed reduction (TPR) is typically used to investigate the redox properties of perovskite oxides. Generally speaking, the lower temperature of the TPR peak, the better reducibility of the perovskite catalyst. As shown in Fig. 3a, selective-dissolution LaMnO3 sample exhibited the lowest reduction temperature (305 ℃) among the three samples, suggesting the best low temperature reducibility [23]. Moreover, the surface lattice oxygen is also regarded to be closely related to the reduction reactions [25]. Futai et al. [26] has found the binding energy of surface lattice oxygen corresponds closely to the Tmax of the TPR profile. A lower binding energy of surface lattice oxygen meant easier activation of surface lattice oxygen, and thus a lower Tmax. In return, the difference in reducibility of perovskites can be used to determine the activity of surface lattice oxygen in a sense, which significantly influences the catalytic performance of catalysts. Regulating the redox properties of perovskites would be plausible for the design of highly active perovskite catalysts. Partial substitution of A-site cation (A1-xA'xBO3) not only forms a less crystallized perovskite structure easier to be reduced, but also contributes to generation of high content of B-site cations in high valence due to principle of electrical neutrality. The increases of oxidation state and B-cation content would promote the reactivity of oxygen adjacent to B cations and thus significantly improve the low-temperature reducibility of perovskites. For substitution of B-site cations, there seems no obvious superiority for the enhancement of catalytic activity compared to A-site substitution although redox properties are mainly related with B-site cations. Therefore, the partial substitution of A-site cations is an extremely effective strategy to improve the redox properties of catalysts and thus enhance their catalytic activity. Up to now, however, it is still very hard to sum up a convincing principle for the selection of doped cations toward better redox property.

|
Download:
|
| Fig. 3. (a) H2-TPR profiles of the selective-dissolution (SD) LaMnO3, three dimensionally ordered macroporous (3DOM) LaMnO3 and γ-MnO2 samples; (b) O2-TPD profiles of the selective-dissolution (SD) LaMnO3, three dimensionally ordered macroporous (3DOM) LaMnO3 and γ-MnO2 samples. Copied with permission [23]. Copyright 2015, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
3.2. Design of oxygen mobility
The oxygen mobility is also invoked as another important factor in catalytic performance of perovskite oxides [27, 28]. It was reported that different oxygen species can be generated/transformed in perovskite oxides accompanied with the formation of oxygen vacancies. Various oxygen species could adsorb on the perovskite oxides, while only two types of oxygen desorb from the surface. Generally speaking, the desorbed oxygen at low temperatures ( < 400 ℃) are assigned as surface adsorbed oxygen (O-, O2-, O22-), which weakly bounded to the oxides surface. The desorbed oxygen species above 400 ℃ are ascribed to lattice oxygen (O2-) in the oxides lattice [23, 29, 30]. In Fig. 3b, the lower desorption temperature of lattice oxygen for the selectivedissolution (SD) LaMnO3 catalyst suggested that the sample had better lattice oxygen mobility [23]. In addition, the high density of oxygen vacancies in perovskite crystals can contribute to the flexible accommodation of highly mobile oxygen, and hence significantly enhance the catalytic performance. By a rational substitution of A or B cations, oxygen vacancies could also be generated (ABO3-δ) in the crystal structure to increase the oxygen mobility, thus facilitating catalytic reactions. For instance, it was reported that Ce doping in A site could slightly enhance interfacial activity due to the increased oxygen mobility. And mixed B-metal composition (substitution by Co) in lanthanum ferrite catalysts (La1-xCexCo1-yFeyO3-δ) could also help modulate both oxygen mobility and catalyst stability. Furthermore, engineering an oxygen defect by physio-chemical means (such as high temperature and high pressure treatment [31] and solution corrosion [32]) could also contribute to the enhancement of oxygen mobility. As mentioned above, the first C-H bond activation and breakage is the rate-determinant step in methane catalytic combustion. Ren et al. [33] has found that the enhanced oxygen mobility is beneficial to promote C-H activation and thus improves the reaction kinetics. The high reducibility and oxygen mobility at low temperatures are two key parameters for high catalytic activity of perovskites, which could hint us towards rational design of goal-oriented catalysts.
3.3. Design of nanoscale structureMorphology of nanostructured perovskite catalysts is also closely associated with the catalytic activity. Different architectural characteristics would bring about the difference in physicochemical properties. Liu et al. [34] prepared ultra-fine nanoparticles of La1-xAxMnO3 (A = Ba, Sr or Ca) with high surface area (29.7–47.0 m2/g), which exhibited high catalytic activity for CH4 total oxidation. Li et al. [35] synthesized one-dimensional single-crystalline nanowires of perovskite oxides, La0.5Sr0.5CoO3, via a dextrose-assisted hydrothermal route, which showed outstanding performance for methane combustion (T50 = 461 ℃). The good catalytic performance of the perovskites catalysts was attributed to the high surface area (17.7 m2/g) of perovskites, as well as a high surface oxygen concentration and the superior lowtemperature reducibility. In addition, it was worth mentioning that Wang et al. has found that two-dimensional layered perovskite La3Mn2O7+δ exhibited superior activity and stability compared to the conventional perovskite LaMnO3. That was because layered La3Mn2O7+δ possessed abundant and stable active lattice oxygen species [36]. The three-dimensionally assembled perovskite nanostructure with ordered channel/pores are also reported as efficient catalysts for methane combustion. The porous skeleton of mesopore-structure and macropore-structure contributes to the enhancement of the surface area, which is directly related to the reaction rate in hydrocarbon combustion [37]. Li et al. [38] prepared three-dimensionally ordered macroporous La0.6Sr0.4MnO3 using polymethyl methacrylate (PMMA) microspheres as a hard template. The three-dimensional-oriented nanostructure perovskites possessed extremely higher specific surface area (32–40 m2/g) than that of bulk La0.6Sr0.4MnO3 (2.6 m2/g). And the highly ordered and interconnected macropores would contribute to the effective diffusion of gas molecules. Furthermore, the well-defined structure and geometrical configurations of mesoporous and macroporous perovskite oxides-based catalysts are greatly beneficial for the rapid and abundant contact of the gas-solid phase, thus improving the reactants transport and materials utilization efficiency. The increased exposure of active sites on ordered pore structure can, therefore, give rise to enhanced catalytic performance of catalysts. One dimensional (1D) perovskite oxides with nano-array structure is regarded promising for catalytic purification of pollutant gases due to the ordered macroporous channels of these nanoarray structures and regular mesoporous structure formed by the gap of LSCO nanoparticles [39-41]. All of these could provide useful guidance for rational design of high-performance catalysts towards low temperature catalytic combustion of methane.
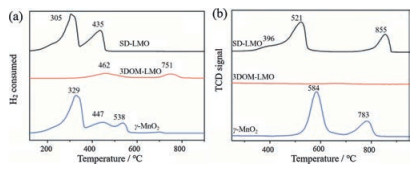
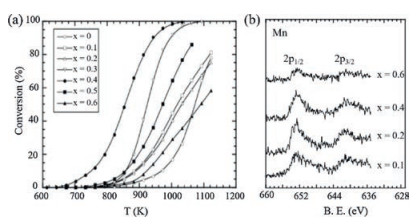
4. Recent advances of perovskite oxides for catalytic combustion of methane 4.1. Perovskite oxides of A/B-site substitutionAs mentioned above, the B-site cations correlates strongly with the catalytic performance of perovskite oxides due to their intrinsic redox properties. So substituting part of B-site cations with other transition metal cations could also modify the redox properties of perovskite oxides, thus enhancing catalytic activity. Ca(Mn1-xTix) O3-δ, synthesized by Taguchi et al. via a sol-gel method, exhibited the highest catalytic activity of CH4 oxidation at x = 0.4 [42]. Fig. 4a showed the light-off curve of CH4 combustion over a range of substituted perovskites. Due to the Mn3+ site was the adsorption site of O2, the author attributed the enhanced catalytic activity to the increased content of Mn3+ on perovskite oxides surface induced by Ti dopig in B-site (Fig. 4b). Najjar et al. [43] studied the effect of aluminum incorporation into the B sublattice of lanthanum manganite on the thermal stability and catalytic activity in CH4 complete oxidation. They found LaMn0.9Al0.1O3+δ exhibited the best performances in terms of activity and stability on stream due to the high mobility and availability of oxygen combined with a facile Mn redox. Huang et al. [44] prepared Fe-substituted hexaaluminates (LaFexAl12-xO19, x = 0-2) and they found LaFeAl11O19 catalyst exhibited even higher activity than conventional Pd/Al2O3 catalyst. The enhanced activity was related to the Fe3+ doped in both Al (3) and Al (5) sites in the mirror planes. La(Fe, B)O3, prepared by substituting Fe with B, also showed better catalytic performance than undoped LaFeO3. Yoon et al. [45] studied the effect of different ions (Mn, Co, Fe) substitution in B sublattice of single phase perovskite for methane oxidation. Feand Co-doped perovskites showed better catalytic activity for the methane oxidation than Mn-doped powder, and the authors proposed that it may be associated with the high ratio of Fe3+/Fe4+ or Co3+/Co4+ compared to Mn3+/Mn4+. Therefore, it is clear that Bsite substitution will influence the chemical state of B-site cations and potentially lead to high-activity perovskite catalysts.

|
Download:
|
| Fig. 4. (a) The light off curve of CH4 combustion on Ca(Mn1-xTix)O3-δ; (b) The X-ray photoelectron spectroscopy spectra of the Mn 2p level in Ca(Mn1-xTix)O3-δ. Copied with permission [42]. Copyright 2007, John Wiley and Sons. | |
Although the A-site cations in perovskite are generally not directly involved in redox process and mainly influence thermal stability of perovskite oxides, substitution of A-site cation is also an efficient technique to modulate catalytic activity. The A-site cations substitution with different valent ions would generate cation vacancies, resulting in the formation of defective perovskites. Liu et al. [34] demonstrated that the ultra-fine particles of La1-xBaxMnO3 by Na2CO3-NaOH coprecipitation method had much higher surface area and better thermal stability than LaMnO3. More importantly, the so-prepared La0.8Ba0.2MnO3 exhibited higher catalytic activity for CH4 total oxidation, with 100% CH4 conversion at 390 ℃, significantly lower than that (420 ℃) of LaMnO3. A portion of Mn3+ would transform into Mn4+ and form highly oxidative oxide as the result of Ba2+ incorporation, which contributed to high catalytic activity of La1-xBaxMnO3. Due to the matched atomic size, A-site cations substitution with strontium (Sr) has been widely studied by researchers [21]. Ponce et al. [13] prepared perovskite-type oxides La1-xSrxMnO3 (x = 0-0.5) by the amorphous citrate process. The authors found that La0.8Sr0.2MnO3 perovskite showed the highest intrinsic catalytic activity at 20% substitution (x = 0.2), which was possibly due to the high stability of Mn4+. However, the intrinsic link between high-oxidation state cations and the enhanced catalytic activity is still unrevealed. Najjar et al. [46] had investigated the effect of praseodymium and europium doping in La1-xLnxMnO3+δ (Ln: Pr or Eu, 0 ≤ x ≤ 1) perovskite catalysts for total methane oxidation. La0.8Eu0.2MnO3.11 showed the highest catalytic activity for methane combustion and the stability of Mn4+ in this catalyst seems to be one of the most important determining factors in the enhanced activity. It was reported that Zr4+ cations tended to occupy the tetrahedral A site of perovskite crystal structure. Li et al. [47] studied the effect of tetrahedral A site substitution with tetravalent (Zr) metal ions over CoCr2O4 for methane catalytic combustion performance. The Co0.95Zr0.05Cr2O4 catalyst exhibited the most active catalytic performance with 90% of methane conversion at 448 ℃, which was 66 ℃ lower than that of the undoped CoCr2O4 catalyst. The doping of zirconium substitution in A-site cations enhances the reducibility of oxides and decreases the bonding strength of Co-O bond and the Cr-O bond, which significantly contributes to the improvement of catalytic activity toward methane combustion. Pure ferrites AFeO3 are rarely investigated due to their relatively low stability. Pecchi et al. [48] demonstrated that the doping of a moderate amount of Ca in A-site cation could improve the oxidation activity due to the generation of anionic vacancies and an over oxidation of Fe3+ to Fe4+. Zhao et al. [49] investigated substitution of La by Ce in LaFeO3 and showed La0.7Ce0.3FeO3 exhibited much higher oxidative performance than pure LaFeO3 for methane combustion. They thought introduction of Ce4+ ion led to stronger interactions with adsorbed O2 and the O-O bond is activated, thus resulting in the enhancement of activity. Double perovskites associating Ni and Al were prepared by Hu et al. [50], who showed that Sr doped double perovskite La1.9Sr0.1NiAlO6 catalyst exhibited significantly improved catalytic activity compared with that of La2NiAlO6.
Substitutions of either B or A cations have been proven to be an effective technique for enhancing catalytic activity of the perovskite oxides for methane combustion. But the underlying rational substitution principle for A/B site is still not clear.
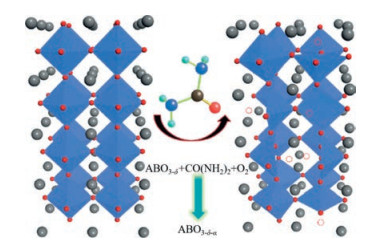
4.2. Perovskite oxides of oxygen defectsBesides the reducibility based on B site elements, oxygen vacancies also play important role in the surface oxidation of CH4. The formation of oxygen vacancies facilitates the enhancement of the mobility of lattice oxygen and dramatically improves the perovskites catalytic activity. Fierro et al. prepared oxygendeficient SrFeO3-δ (0.02 < δ < 0.26) perovskites by soft-chemistry procedures to investigate the effect of the oxygen vacancies amount on methane oxidation activity [51]. They observed that the highly deficient SrFeO2.74 exhibited the maximum catalytic activity for methane combustion. The highest content and ordered arrangement of oxygen vacancies were regarded as the key feature for the enhanced catalytic performance of the most oxygendeficient oxides SrFeO2.74. The study about deficient oxides suggested the presence of O vacancies in perovskite oxide indeed play a positive role on methane combustion. As we know, the largest amount of O vacancies in SrFeO2.74 may significantly increase oxygen mobility and thus improve its catalytic activity. Recently, Feng et al. [32] developed a defect engineering route to fabricate oxygen-deficient perovskite oxide (La0.5Sr0.5CoO3-δ), as shown in Fig. 5. They adopted solid topochemical reactions to regulate the concentration of oxygen vacancies and octahedral distortions by adjusting the mass ratio of La1-xSrxCoO3 and urea. Urea pyrolysis was able to increase the oxygen defect concentration of perovskite oxides and enhance oxygen mobility, thus contributing to improve the catalytic activity of CO oxidation. In addition, substitution of A/B site cations of perovskites could also contribute to the formation of oxygen vacancies due to the oxide stoichiometry and electrical neutrality principle, especially for substitution of A-site ions. Fierro et al. [52] found the partial substitution of La by Sr (La1-xSrxNiO3) would result in the formation of a mixture of NiⅡ/NiⅢ oxidation states and oxygen vacancies, remarkably improving the oxygen mobility and thus enhancing the intrinsic activity for the CH4 combustion. Inspired by that, defect engineering over perovskite oxide may provide a new avenue to design highly efficient catalysts for CH4 combustion.

|
Download:
|
| Fig. 5. Scheme of synthetic approach for oxygen-deficient perovskite La1-xSrxCoO3; the red spheres represent oxygen atoms; gray spheres represent La/Sr atoms; white spheres with red dotted lines represent oxygen vacancies. Copied with permission [32]. Copyright 2017, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
4.3. Porous perovskite oxides
The textural characteristics are proved to be a critical factor for the perovskite catalytic activity, since physical textural properties are usually involved with surface area increase and active sites exposure. To date, many efficient synthesis routes, such as nonaqueous solvothermal synthesis route, self-propagating high-temperature synthesis, mesostructuration and macrostructuration approaches, have been investigated to optimize specific surface areas and internal physical structure of perovskites. Aruna et al. [53] prepared Ln1-xSrxMnO3(Ln = Pr, Nd, Sm) powders after the ignition at 300 ℃ by auto combustion process, which presented surface areas in the range of 13–40 m2/g. Furthermore, auto combustion process was observed to lead to excess Mn4+, facilitating the catalytic reaction.
Mesoporous materials with well-defined porosity provides a novel strategy for engineering perovskites textural structure due to the revolution in nanoscience. Nanocasting is a versatile route to obtain oxide materials with organized mesopore. Wang et al. [54] prepared ordered mesoporous LaCoO3 perovskite oxide by using ordered mesoporous cubic vinyl silica as the hard template for the first time, which presented a high surface area of 96.7 m2/g. The mesoporous perovskite oxides LaCoO3 exhibited excellent catalytic performance for CH4 combustion. The light-off temperature (T10) and the half-conversion temperature (T50) of mesoporous LaCoO3 were at 335 ℃ and 470 ℃, respectively, which was 165 ℃ and 125 ℃ lower than that of bulk perovskite oxides prepared by the conventional citrate method (Fig. 6a). The authors have attributed its outstanding catalytic activity to high valent cobalt ions and high content of O22-/O-, species existed in mesoporous LaCoO3 (Figs. 6b and c). Zhang et al. [55] proposed that the presence of high-valence state cations was beneficial for the desorption of oxygen at low temperatures. And the high-valence state and high content of B cations would enhance the reactivity of oxygen bonded to B cations. Therefore, more active oxygen species and a greater amount of active sites are exposed in the high-surface-area mesoporous oxides surface, which facilitates higher catalytic performance. However, it should be noted that a slight decrease (to 69.5 m2/g) in surface areas of mesoporous LaCoO3 has been observed after the first methane activity test, which may be due to that a small fraction of particles have sintered during the catalytic reaction. The slight textural disruption occurring during reactions should be alerted because it is closely relevant to the durability of catalysts. Nie et al. [56] also prepared meso/macroporous NiO/ LaNiO3 composite catalysts by a combined citric acid combustion, which showed enhanced methane catalytic oxidation performance due to the higher surface area than LaNiO3 obtained only by a citric acid combustion method. Furthermore, the formation of NiO particles on the catalysts surface is beneficial for the activation of the C-H bond, thus further contributing to facilitating catalytic reaction. The well-defined pore structure of mesoporous catalysts is a key parameter for their stable activity. Ensuring the stability of the mesoporous structure is a critical issue, which is what we need to focus on in the future.

|
Download:
|
| Fig. 6. (a) Temperature dependence of methane combustion on (δ1) mesoporous LaCoO3 after removal of silica, (δ2) mesoporous LaCoO3/silica hybrid material and (δ3) conventional LaCoO3, and (δ4) used mesoporous LaCoO3 catalyst; (b) XPS spectra of the Co 2p and O 1s levels of the mesoporous and conventional LaCoO3. Reproduced with permission [54]. Copyright 2008, American Chemical Society. | |
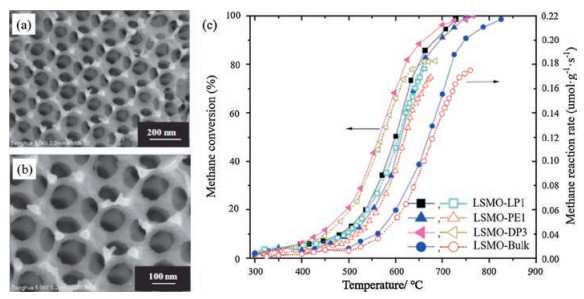
Over the past years, a unique strategy has been proposed and developed for fabricating three-dimensionally interconnected macroporous walls involving introduction of regularly-aligned colloid crystals templates [57]. Inspired by that, one can create a three-dimensionally macroporous (3DOM) structure catalysts using the colloidal crystal templating method mentioned above to prevent the destruction of pore structures and decrease of associated surface areas. Li et al. have fabricated three dimensionally ordered macroporous rhombohedral La0.6Sr0.4MnO3 (3DOM LSMO) for the catalytic combustion of methane by the surfactantassisted colloidal crystal polymethyl methacrylate-templating strategy [38]. Introduction of a surfactant was beneficial for the generation of the 3DOM architectures on the skeletons. They found that addition of appropriate amounts of dimethoxytetraethylene glycol and polyethylene glycol was beneficial for preparation of high-quality 3DOM-structured La0.6Sr0.4MnO3 shown in Figs. 7a and b, which had great significance for improving stability of macroporous structure. Fig. 7c showed the catalytic activities of LSMO samples with the addition of different amounts of surfactants. The 3DOM La0.6Sr0.4MnO3 catalyst (labeled LSMODP3) derived with the proper surfactant (3.0 mL dimethoxytetraethylene glycol, 5.0 mL polyethylene glycol and 9.0 mL water) exhibited an excellent catalytic activity for methane combustion. T10, T50 and T90 (abbreviation of 10%, 50% and 90% conversion temperature) were 437 ℃, 566 ℃, and 661 ℃, respectively. The high oxygen species concentration, high surface area and superior low-temperature reducibility should be responsible for its great catalytic activity for CH4 combustion. In addition, Arandiyan et al. [58] also fabricated newly designed 3D highly macro/mesoporous multifunctional La1-xCexCoO3 nanohybrid frameworks with a 2D hexagonal mesostructure by facile meso-molding in a threedimensionally macroporous perovskite (MTMP) route. The highsurface-area monolithic nanohybrid 3DOM-m La1-xCexCoO3 catalysts exhibited much higher catalytic activity for methane combustion compared with bulk one dimensionally disordered nonporous (1DDN) La1xCexCoO3 (0 > x > 0.9) samples. Especially, 3DOM-m La0.7Ce0.3CoO3 catalyst with the lowest apparent activation energy (53.3 kJ/mol) performed the best with T10, T50, and T90 at 381 ℃, 479 ℃ and 555 ℃, respectively, which is due to that it possessed a larger surface area/pore volume, uniform pore sizes, higher accessible surface oxygen concentration, better lowtemperature reducibility, and a unique nanovoid 3D structure. It is worth mentioning that Wang et al. [59] prepared a mesostructured 3D network composing of hexapod-shaped multi-metal oxide crystalline nanoparticles by disassembling an ordered porous La0.6Sr0.4MnO3 perovskite array, which exhibited excellent low temperature methane oxidation activity (T90 = 438 ℃). According to their results, the enhanced activity was linked with a modified mesoporous structure, a richness of surface oxygen species, better lattice oxygen mobility and exposure of the (001) facet along the fractured faces. This new strategy dramatically provides exciting possibilities for attaining highly effective materials for catalytic systems.

|
Download:
|
| Fig. 7. (a), (b) SEM images of La0.6Sr0.4MnO3 (LSMO)-DP3; (c) Methane conversion and methane reaction rate versus temperature over the LSMO-LP1, LSMO-PE1, LSMO-DP3, and LSMO-bulk catalysts. DP3, LP1 and PE1 represents different content and different types of surfactant. Reproduced with permission [38]. Copyright 2013, Elsevier Inc. | |
To further enhance catalytic performance of 3DOM perovskite based catalysts, highly dispersed noble metal nanoparticles supported on high-surface area 3DOM perovskite framework were successfully generated [60-64], which showed super catalytic performance for methane combustion. Previously, Arandiyan et al. had successfully synthesized high-surface-area Ag/3DOM La0.6Sr0.4MnO3 catalysts with controlled Ag nanoparticle sizes [60, 61], which indeed exhibited excellent catalytic performance due to its larger surface area, higher oxygen ad species concentration, better low-temperature reducibility, and unique nanovoid 3DOM structure. Xu et al. [62] had also prepared threedimensionally ordered macroporous (3DOM) LaMnAl11O19-supported Pd nanocatalyst, which also showed much better catalytic activity for methane combustion than that of 3DOM LaMnAl11O19. After that, three-dimensionally ordered macroporous (3DOM) La0.6Sr0.4MnO3 (LSMO) perovskite catalysts with bimetallic Au-Pd alloy nanoparticles (NPs) dispersed on nanohybrid were first fabricated by Arandiyan et al. [63] via the L-lysine-mediated colloidal crystal templating and reduction routes. The bimetallic catalyst Au-Pd/3DOM LSMO with well-dispersed Au-Pd alloy NPs (2.05–2.35 nm in size) exhibited excellent catalytic activity (T10 = 265 ℃, T50 = 314 ℃, and T90 = 336 ℃) as well as higher reaction rate compared with single 3DOM LSCO catalyst. A richness of adsorbed oxygen species, increased oxidized noble metal species, low-temperature reducibility, and strong noble metal-3DOM LSMO interaction were responsible for their enhanced activity. Furthermore, Au-Pd/3DOM LSMO sample also exhibited superior hydrothermal stability, which was of great significance to access industrial application potential. Dai et al. [64] also prepared 3DOM CoFe2O4 with the co-load of MnOx and Pd-Pt alloy nanoparticles, and they showed the dispersion of Pd-Pt NPs on 6.70 wt% MnOx/3DOM CoFe2O4 (6.70 wt% means the loading weight percentage of MnOx on CoFe2O4 is 6.70%) significantly enhanced the catalytic performance compare with the unsupported 3DOM CoFe2O4. However, it should be noted that the introduction of SO2 would result in the reversible deactivation of these precious metal supported catalysts. Dai et al. also prepared a series of high-surface-area and high-quality-framework Au/3DOM LaCoO3 [65], Au/3DOM La0.6Sr0.4MnO3 [66], 3DOM LaMnO3 [67] and 3DOM Eu0.6Sr0.4FeO3 [68] by adopting the colloidal crystal templating (CCT) technique, which exhibited superior catalytic performance toward organic oxidation. It has been proven that the morphology of materials is relevant to their catalytic performance, because the amount of exposed crystal planes with high activity is different in variously-morphological materials [11, 69]. Therefore, promising catalysts with controlled physical morphology and superior stability for methane or other hydrocarbon combustion is desired.
4.4. Nano-array perovskite oxides catalystsAs mentioned above, morphology of nanostructured perovskite catalysts is closely related to their catalytic activity. The welldefined mesoporous and macroporous structure of perovskite oxides-based catalysts contribute to fast and abundant gas diffusion, thus improving catalytic reaction efficiency. The configured nano-array perovskites catalysts could also achieve an efficient gas diffusion and catalytic reaction due to the ordered macroporous channels of the array structures.
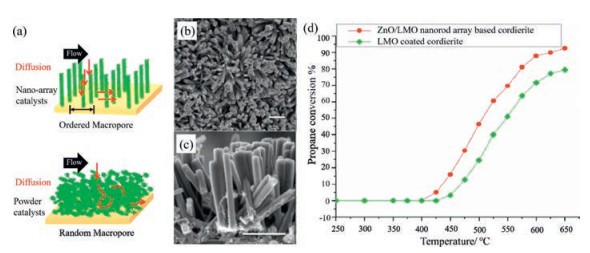
The traditional particle-form perovskite-based monolith catalysts fabricated by empirical wash-coating processes exhibit compromised catalytic activity at low temperatures due to their disordered pore structure, random active sites and poor washcoat adhesion. Recently, the ordered nano-array perovskite catalysts have been proposed to mitigate these issues. Fig. 8a presented a schematic transport model of nano-array catalysts and traditional nano-powder catalysts [70]. The well-defined structural and geometrical configurations not only exposed more active sites and improved material utilization efficiency, but also promoted the gas-solid interaction by a much shorter diffusion length and a more direct path, which enabled heterogeneous reactions activity enhanced significantly. Guo et al. [70] have firstly fabricated a broad spectrum of metal oxide based monolithic nano-array catalysts by directly integrating bare monolith structures with ultra-efficient, thermally stable, and physically and chemically well-defined nanostructure arrays. The morphology of metal oxide nano-array could be tuned from nanorod, nanotube to mesoporous nanowire. As they expected, activity evaluation results demonstrated superior catalytic performance of nano-array configuration over the conventional wash-coated catalysts. Furthermore, the catalytic activity could also be well controlled by tuning the geometrical morphology of nano arrays. Accordingly, the rational design of nano-array structure of catalysts may provide a favorable direction for developing high-activity and cost-effective perovskite catalysts.

|
Download:
|
| Fig. 8. (a) A schematic transport model of nano-array catalysts and traditional nano-powder catalysts. Reproduced with permission [70]. Copyright 2013, Elsevier Ltd. SEM images of (b) top view and (c) cross-sectional view of ZnO/ZnO/LaMnO3(LMO) core-shell nanorod arrays; (d) Catalytic performance of powder-form LMO based and ZnO/LMO core–shell nanorod arrays based catalysts for propane oxidation. Scale bars without labels are 1 mm. Reproduced with permission [39]. Copyright 2015, Elsevier B.V. | |
Guo et al. [71] have fabricated vertically-aligned TiO2/(La, Sr) MnO3 composite nanorod array catalysts using a combination of wet chemistry synthesis and physical sputtering (radio-frequency magnetron sputtering). The TiO2/(La, Sr)MnO3 core-shell nanorod arrays catalysts exhibited drastically enhanced CO oxidation activity compared to TiO2 nanorods, which provided a strategy model for the design of high-activity nano-array perovskite catalysts. Wang et al. have successfully fabricated ZnO/perovskite (LaBO3, B = Mn, Co, Ni) core-shell nanorod arrays for hydrocarbon oxidation within three-dimensional (3D) honeycomb cordierite substrates by a hydrothermal synthesis combined with colloidal deposition methods [39]. In brief, dispersed perovskite nanoparticles were uniformly coated onto the large scale ZnO nanorod arrays rooted on the substrates to obtain the core-shell ZnO/ perovskite composites. Figs. 8b and c displayed the top-view and cross-sectional view of ZnO/LaMnO3(LMO) core-shell nanorods array catalysts, which exhibited well-aligned and densely packed array characteristics. The nanorods arrays distributed inside the substrate evenly with length of 1.5 μm and diameter of 100–200 nm. And the ZnO/LMO composited catalysts exhibited an enhanced catalytic performance for propane oxidation with 25 ℃ lower light-off temperature than powder-form washcoated perovskite catalyst of similar LaMnO3 loading (4.3 mg) (Fig. 8d). They attributed the improved catalytic activity of nano-array catalysts to the good dispersion, size control in La-based perovskite nanoparticles and their interfaces between perovskite and ZnO. Ren et al. [33] had reported that the monolithically integrated MxCo3-xO4 (M = Co, Ni, Zn) nanoarrays-based catalysts exhibited tunable catalytic performance towards CH4 combustion. The Ni0.5Co2.5O4 nano arrays reached total oxidation for methane below 600 ℃. The macro sized perovskite-like oxide La1-xSrxCuO4 (x = 0, 1) single crystallites with a rod-like morphology were prepared by He et al. [72], and LaSrCuO4 catalyst exhibited good catalytic performance for methane oxidation (T50 = 620 ℃). Li et al. [35] synthesized one-dimensional single-crystalline nanowires of perovskite oxides, La0.5Sr0.5CoO3, which showed outstanding performance for methane combustion (T50 = 461 ℃). Our recent research on methane combustion over perovskite nano-array catalysts also showed a promising performance. Therefore, the introduction of nano-array perovskite catalysts may provide a plausible avenue for methane catalytic combustion at low temperatures. In a word, the emerging nano-array structure perovskites catalysts opens up numerous possibilities to improve their performance based on broad perovskites families. We could not only control and manipulate the nano-array morphology and support structures, but also tune the composition of perovskites to achieve the perfect combination scheme.
As a brief summary, all the reviewed perovskite oxide nanocatalysts and their catalytic performance for methane combustion are listed in Table 1.
|
|
Table 1 Activity of perovskite based catalysts for methane combustion. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5. Summary and perspectives
In this review, we have summed up recent research progress on perovskites catalysts for methane combustion. The high activation energy of the first C-H bond in CH4 molecule has further hampered the methane catalytic combustion at low temperature. The traditional particle-form perovskite catalysts exhibit compromised catalytic performance due to their low surface area, random active sites and inadequate gas-solid interaction. Researchers have made many subsequent attempts to mitigate and address these issues. Reducibility, lattice oxygen activity and oxygen mobility are essential for the methane catalytic combustion on perovskite surface via an MvK mechanism. Therefore, nano-structure perovskites catalysts with A/B-site substitution, defect engineering and various morphology such as nanoparticle, nanowire and layered nanostructure were fabricated to magnify the above mentioned surface property. Besides, the three-dimensional ordered nanostructure design including ordered mesoporous/ macroporous structure and nano-array structure design has been proved to be a promising strategy of high-activity perovskite catalysts. The unique morphology and ordered porous structure enabled much bigger surface area and more exposure of active sites, thus significantly enhancing the catalytic activity. Meanwhile, the monolithic nano-array perovskite catalysts with welldefined facet and ordered porous channel exhibited excellent catalytic performances for hydrocarbon combustion. We believed nano-structured perovskites oxides would be a promising highperformance, non-precious metal catalysts for CH4 combustion.
To date, great progress on perovskite-based catalysts for methane catalytic combustion has been reached. While there are still several issues need to address in the future to fabricate robust and high-performance perovskites-type catalysts: (1) The general principle of A/B substitution effect on redox property and oxygen mobility is still unclear. More experimental effort is needed to provide a regular principle for perovskites catalysts rational design; (2) The quantitative relationship between surface adsorbed oxygen, oxygen vacancies and surface lattice oxygen mobility, which is crucial for high activity catalysts design, should be further explored; (3) The two-dimensionally (2D) layered perovskite materials exhibits superior activity and stability for heterogeneous catalysis, as they can maximize the exposure of active sites and to accommodate reactants in abundance. Therefore, the quest for a feasible approach of fabrication and rational design of 2D perovskite oxides is extremely necessary; Activity degradation-induced structure collapse has hindered the wide application of nanostructured porous perovskite catalysts. Fabrication of robust ordered porous perovskites catalysts is an important challenge need to be solved.
AcknowledgmentsThe authors are grateful for the financial support from the Recruitment Program of Global Young Experts Start-up Fund and the Program of Introducing Talents of Discipline to Universities of China (111 Program, No. B17019).
| [1] |
H. Herzog, D. Golomb, Encycl. Energy 1(2004) 1-11. |
| [2] |
X. Li, Y. Liu, J. Deng, Y. Zhang, et al., Appl. Surf. Sci. 403(2017) 590-600. DOI:10.1016/j.apsusc.2017.01.237 |
| [3] |
F.F. Tao, J. Shan, L. Nguyen, et al., Nat. Commun. 6(2015) 1-9. |
| [4] |
N.M. Kinnunen, J.T. Hirvi, K. Kallinen, et al., Appl. Catal. B:Environ. 207(2017) 114-119. DOI:10.1016/j.apcatb.2017.02.018 |
| [5] |
E. Campagnoli, A.C. Tavares, L. Fabbrini, et al., J. Mater. Sci. 41(2006) 4713-4719. DOI:10.1007/s10853-006-0057-0 |
| [6] |
L.L. Smith, H. Karim, M.J. Castaldi, S. Etemad, W.C. Pfefferle, Catal. Today 117(2006) 438-446. DOI:10.1016/j.cattod.2006.06.021 |
| [7] |
Y. Wang, M. Guo, J. Lu, M. Luo, Chin. J. Catal. 32(2011) 1496-1501. |
| [8] |
W.R. Schwartz, D. Ciuparu, L.D. Pfefferle, J. Phys. Chem. C 116(2012) 8587-8593. DOI:10.1021/jp212236e |
| [9] |
V. Viitanen, H. Jiang, M. Honkanen, et al., Top. Catal. 56(2013) 576-585. DOI:10.1007/s11244-013-0017-2 |
| [10] |
J. Li, J. Zhang, Z. Lei, B. Chen, Energy Fuels 26(2012) 1-8. DOI:10.1021/ef2018786 |
| [11] |
Z. Ren, Y. Guo, P.X. Gao, Catal. Today 258(2015) 448-453. |
| [12] |
J. Deng, L. Zhang, H. Dai, H. He, C. Tong, J. Mol. Catal. A:Chem. 299(2009) 60-67. DOI:10.1016/j.molcata.2008.10.006 |
| [13] |
S. Ponce, M.A. Peña, J.L.G. Fierro, Appl. Catal. B:Environ. 24(2000) 193-205. DOI:10.1016/S0926-3373(99)00111-3 |
| [14] |
J. Niu, J. Deng, W. Liu, et al., Catal. Today 126(2007) 420-429. DOI:10.1016/j.cattod.2007.06.027 |
| [15] |
Y.H. Chin, C. Buda, M. Neurock, E. Iglesia, J. Am. Chem. Soc. 135(2013) 15425-15442. DOI:10.1021/ja405004m |
| [16] |
Y. Feng, P.M. Rao, D.R. Kim, X. Zheng, Proc. Combust. Inst. 33(2011) 3169-3175. DOI:10.1016/j.proci.2010.05.017 |
| [17] |
Y. Zhang, Z. Qin, G. Wang, et al., Appl. Catal. B:Environ. 129(2013) 172-181. DOI:10.1016/j.apcatb.2012.09.021 |
| [18] |
M.A. Peña, J.L.G. Fierro, Chem. Rev. 101(2001) 1981-2018. DOI:10.1021/cr980129f |
| [19] |
R. Auer, F.C. Thyrion, Ind. Eng. Chem. Res. 41(2002) 680-690. DOI:10.1021/ie0104924 |
| [20] |
F. Zasada, J. Janas, W. Piskorz, M. Gorczynska, Z. Sojka, ACS Catal. 7(2017) 2853-2867. DOI:10.1021/acscatal.6b03139 |
| [21] |
H. Zhu, P. Zhang, S. Dai, ACS Catal. 5(2015) 6370-6385. DOI:10.1021/acscatal.5b01667 |
| [22] |
J. Zhu, A. Thomas, Appl. Catal. B:Environ. 92(2009) 225-233. DOI:10.1016/j.apcatb.2009.08.008 |
| [23] |
W. Si, Y. Wang, Y. Peng, J. Li, Angew. Chem. Int. Ed. 54(2015) 7954-7957. DOI:10.1002/anie.201502632 |
| [24] |
W. Tang, X. Wu, D. Li, et al., J. Mater. Chem. A 8(2014) 2544-2554. |
| [25] |
L. Hu, K. Sun, Q. Peng, B. Xu, Y. Li, Nano Res. 3(2010) 363-368. DOI:10.1007/s12274-010-1040-2 |
| [26] |
F. Ma, Y. Chen, H. Lou, Kinet. Catal. Lett. 31(1986) 47-53. DOI:10.1007/BF02062510 |
| [27] |
S. Royer, D. Duprez, S. Kaliaguine, J. Catal. 234(2005) 364-375. DOI:10.1016/j.jcat.2004.11.041 |
| [28] |
M. Alifanti, J. Kirchnerova, B. Delmon, D. Klvana, Appl. Catal. A:Gen. 262(2004) 167-176. DOI:10.1016/j.apcata.2003.11.024 |
| [29] |
N. Yamazoe, Y. Teraoka, T. Seiyama, Chem. Lett. 10(1981) 1767-1770. DOI:10.1246/cl.1981.1767 |
| [30] |
T. Nakamura, M. Misono, Y. Yoneda, Chem. Lett(1981), 1589-1592. |
| [31] |
L. Li, G. Li, J. Xiang, R.L. Smith, H. Inomata, Chem. Mater. 15(2003) 889-898. DOI:10.1021/cm020582e |
| [32] |
X. Wang, K. Huang, W. Ma, et al., Chem. Eur. J. 23(2017) 1093-1100. DOI:10.1002/chem.201604065 |
| [33] |
Z. Ren, V. Botu, S. Wang, et al., Angew. Chem. Int. Ed. 53(2014) 7223-7227. DOI:10.1002/anie.201403461 |
| [34] |
Y. Liu, H. Zheng, J. Liu, T. Zhang, J. Chem Eng 89(2002) 213-221. DOI:10.1016/S1385-8947(02)00013-X |
| [35] |
H. Arandiyan, H. Chang, C. Liu, Y. Peng, J. Li, J. Mol. Catal. A:Chem. 378(2013) 299-306. DOI:10.1016/j.molcata.2013.06.019 |
| [36] |
X. Du, G. Zou, Y. Zhang, X. Wang, J. Mater. Chem. A 1(2013) 8411-8416. DOI:10.1039/c3ta11129f |
| [37] |
N. Gunasekaran, S. Saddawi, J.J. Carberry, J. Catal. 111(1996) 107-111. |
| [38] |
H. Arandiyan, H. Dai, J. Deng, et al., J. Catal. 307(2013) 327-339. DOI:10.1016/j.jcat.2013.07.013 |
| [39] |
S. Wang, Z. Ren, W. Song, et al., Catal.Today 258(2015) 549-555. DOI:10.1016/j.cattod.2015.03.026 |
| [40] |
D. Jian, P.X. Gao, W. Cai, et al., J. Mater. Chem. 19(2009) 970-975. DOI:10.1039/b817235h |
| [41] |
H.Y. Gao, W.J. Cai, P. Shimpi, H.J. Lin, P.X. Gao, J. Phys. D:Appl. Phys. 43(2010) 1-6. |
| [42] |
H. Taguchi, K. Nakade, M. Yosinaga, M. Kato, K. Hirota, J. Am. Ceram. Soc. 310(2008) 2007-2009. |
| [43] |
H. Najjar, J. Lamonier, O. Mentré, J. Giraudon, H. Batis, Catal. Sci. Technol. 3(2013) 1002-1016. DOI:10.1039/c2cy20645e |
| [44] |
F. Huang, X. Wang, A. Wang, J. Xua, T. Zhang, Catal. Sci. Technol. 6(2016) 4962-4969. DOI:10.1039/C5CY02187A |
| [45] |
J.S. Yoon, Y. Lim, B.H. Choi, H.J. Hwang, Int. J. Hydrogen Energy 39(2014) 7955-7962. DOI:10.1016/j.ijhydene.2014.03.008 |
| [46] |
H. Najjara, H. Batisb, J. Lamoniera, O. Mentréa, J. Giraudon, Appl. Catal. A:Gene 469(2014) 98-107. DOI:10.1016/j.apcata.2013.09.014 |
| [47] |
J. Chen, W. Shi, X. Zhang, et al., Environ. Sci. Technol. 45(2011) 8491-8497. DOI:10.1021/es201659h |
| [48] |
G. Pecchi, M.G. Jiliberto, A. Buljan, E.J. Delgado, Solid State Ionics 187(2011) 27-32. DOI:10.1016/j.ssi.2011.02.014 |
| [49] |
X. Xiang, L. Zhao, B. Teng, et al., Appl. Surf. Sci 276(2013) 328332. |
| [50] |
R. Hu, Y. Bai, H. Du, et al., J. Rare Earth 33(2015) 1284. DOI:10.1016/S1002-0721(14)60558-5 |
| [51] |
H. Falcon, J.A. Barbero, J.A. Alonso, M.J. Mart, J.L.G. Fierro, Chem. Mater. 14(2002) 2325-2333. DOI:10.1021/cm011292l |
| [52] |
R.M. Garcíade la Cruz, H. Falcón, M.A. Peña, J.L.G. Fierro, Appl. Catal. B:Environ. 33(2001) 45-55. DOI:10.1016/S0926-3373(01)00157-6 |
| [53] |
S.T. Aruna, M. Muthuraman, K.C. Patil, Solid State Ionics 120(1999) 275-280. DOI:10.1016/S0167-2738(99)00021-1 |
| [54] |
Y. Wang, J. Ren, Y. Wang, et al., J. Phys. Chem. C 112(2008) 15293-15298. DOI:10.1021/jp8048394 |
| [55] |
H.M. Zhang, Y. Shimizu, Y. Teraoka, N. Miura, N. Yamazoe, J. Catal. 121(1990) 423-440. |
| [56] |
L. Nie, J. Wang, Q. Tan, Catal. Commun. 97(2017) 1-4. DOI:10.1016/j.catcom.2017.04.010 |
| [57] |
J.W. Kim, K. Tazumi, R. Okaji, M. Ohshima, Chem. Mater. 21(2009) 3476-3478. DOI:10.1021/cm901265y |
| [58] |
H. Arandiyan, J. Scott, Y. Wang, et al., ACSAppl. Mater. Interfaces 8(2016) 2457-2463. DOI:10.1021/acsami.5b11050 |
| [59] |
Y. Wang, H. Arandiyan, H.A. Tahini, et al., Nat Commun 8(2017) 15553. DOI:10.1038/ncomms15553 |
| [60] |
H. Arandiyan, H. Dai, J. Deng, et al., Chem. Commun. 49(2013) 10748-10750. DOI:10.1039/c3cc46312e |
| [61] |
H. Arandiyan, H. Dai, J. Deng, et al., J. Phys. Chem. C 118(2014) 14913-14928. |
| [62] |
P. Xu, X. Zhao, X. Zhang, et al., Mol. Catal. 439(2017) 200-210. DOI:10.1016/j.mcat.2017.06.036 |
| [63] |
Y. Wang, H. Arandiyan, J. Scott, et al., ACS Catal. 6(2016) 6935-6947. DOI:10.1021/acscatal.6b01685 |
| [64] |
X. Li, Y. Liu, J. Deng, et al., Appl. Surf. Sci. 403(2017) 590-600. DOI:10.1016/j.apsusc.2017.01.237 |
| [65] |
X. Li, H. Dai, J. Deng, et al., Chem. Eng. J. 228(2013) 965-975. DOI:10.1016/j.cej.2013.05.070 |
| [66] |
Y. Liu, H. Dai, J. Deng, et al., J. Catal. 305(2013) 146-153. DOI:10.1016/j.jcat.2013.04.025 |
| [67] |
Y. Liu, H. Dai, J. Deng, et al., Appl. Catal. 141(2013) 493-505. |
| [68] |
K. Ji, H. Dai, J. Deng, et al., Appl. Catal. B:Environ. 129(2013) 539-548. DOI:10.1016/j.apcatb.2012.10.005 |
| [69] |
X. Xie, Y. Li, Z. Liu, M. Haruta, W. Shen, Nature 458(2009) 746-749. DOI:10.1038/nature07877 |
| [70] |
Y. Guo, Z. Ren, W. Xiao, et al., Nano Energy 2(2013) 873-881. DOI:10.1016/j.nanoen.2013.03.004 |
| [71] |
Y. Guo, Z. Zhang, Z. Ren, H. Gao, P.X. Gao, Catal. Today 184(2012) 178-183. DOI:10.1016/j.cattod.2011.10.027 |
| [72] |
L. Zhang, Y. Zhang, H. Dai, et al., Catal. Today 153(2010) 143-149. DOI:10.1016/j.cattod.2010.02.059 |