 2018, Vol. 29
2018, Vol. 29 



b The State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China;
c Collaborative Innovation Center of Chemical Science and Engineering(Tianjin), Tianjin 300071, China
Over the past two decades, natural cinchona alkaloids and their derivatives have emerged as prevailing chiral organocatalysts and been widely utilized in a growing number of asymmetric organic reactions [1-8]. As homogeneous organocatalysts, cinchona catalysts encounter at least two challenging issues: separation and recycling of the catalyst and the relatively large amount of the catalyst loading. Immobilization of a homogeneous catalyst on a retrievable support is generally considered to be a simple and practical strategy to solve such issues. Heterogenization of cinchona catalysts has therefore attracted significant attention [9-21]. Typical solid supports like polymers, silica have already been utilized to prepare the immobilized cinchona organocatalysts, which can be separated by conventional separation techniques such as filtration and centrifugation.
Recently, magnetic nanoparticles (MNPs) as salient catalyst supports have attracted much attention [22-25]. Owing to their nano-scale nature and high surface area, MNPs-supported catalysts can act in a quasi-homogeneous manner. Also, the magnetic nature of the MNPs support enables the immobilized catalyst to be easily separated from the reaction mixture by simple decantation with the aid of external magnets, instead of tedious centrifugation and filtration. The use of MNPs in the immobilization of cinchona organocatalysts has also drawn some recent interest [26-28]. The 9-amino-9deoxyepiquinine-derived urea, thiourea, and amide organocatalysts [26, 27] and 9-amino-9deoxyepicinchonidine catalyst [28] were successfully immobilized on Fe3O4 MNPs and the corresponding MNPs-supported catalysts were used as recoverable organocatalysts in the asymmetric aldol, Diels-Alder, and Michael reactions with moderate to excellent enantioselectivities achieved. To the best of our knowledge, other MNPs-immobilized cinchona organocatalysts have not been explored yet. In this communication, we report the synthesis of new MNPs-supported cinchona alkaloids such as quinine and quinidine and their application in the asymmetric Michael addition reaction of 1, 3-dicarbonyls and maleimides.
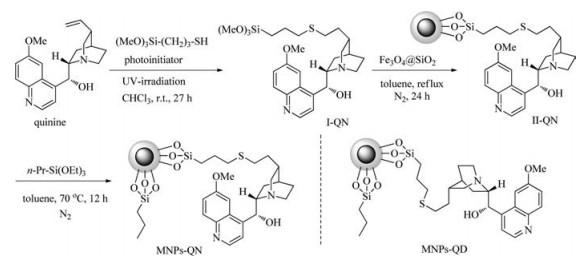
Synthesis of MNPs-supported cinchona alkaloids was illustrated in Scheme 1 (for experimental detail, see Supporting information). Magnetic nanoparticles silica-coated Fe3O4 were readily prepared by the well documented methods [26, 29, 30]. 3-Mercaptopropyltrimethoxysilane was chosen as a preferred linker agent between the MNPs support Fe3O4@SiO2 and cinchona alkaloid. Its anchoring group trimethoxysilyl enables the covalent bonding of the linker on the surface of silica [26]. By the photo-irradiated thio-ene reaction recently developed by Garrell group [31], equimolar quinine and 3-mercaptopropyltrimethoxysilane were smoothly coupled under ultraviolet irradiation at r.t. for 27 h to give the corresponding intermediate Ⅰ-QN in high purity. Intermediate Ⅰ-QN was used in the next step without further purification. Through a sol-gel coupling reaction refluxed in toluene, the quinine-immobilized Fe3O4@SiO2 nanoparticles Ⅱ-QN were readily prepared. Quinine is proven to be a bifunctional organocatalyst in asymmetric reactions including Michael addition reaction, and its hydroxyl group plays a critical role in stereoselectivity through hydrogen bonding interactions [1, 2]. In order to avoid the potential interference of the residual hydroxyl groups on the surface of nanoparticles with the active quinine moiety, the quinine-immobilized Fe3O4@SiO2 nanoparticles were capped with triethoxypropylsilane at 70 ℃ for 12 h. After separation with the aid of an external magnet and washes with dry toluene, the quinine-functionalized Fe3O4@SiO2 nanoparticles (MNPs-QN) were collected as a magnetically retrievable organocatalyst. By the same procedure, the quinidine-functionalized Fe3O4@SiO2 nanoparticles (MNPs-QD) were also smoothly prepared (Scheme 1).

|
Download:
|
| Scheme 1. Synthesis of the catalysts MNPs-QN and MNPs-QD. | |
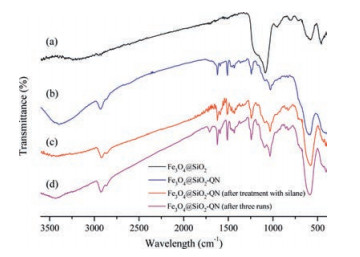
The prepared MNPs-QN was characterized by FT-IR, XPS, SEM measurements, and elemental analysis. IR spectra clearly indicated that quinine was successfully immobilized on the Fe3O4@SiO2 support (Fig. 1). Referring to the IR spectrum of the Fe3O4@SiO2 support (curve a), the IR spectrum of Ⅱ-QN clearly showed the characteristic signals from quinine including C-H stretching at about 2900 cm−1 and quinoline ring vibration at about 1500 cm−1 (curve b). The spectrum of the catalyst MNPs-QN (curve c) unveiled that the broad signal at about 3400 cm−1 assigned to hydroxyl groups substantially diminished. This result confirmed that treatment with triethoxypropylsilane effectively scavenged the residual hydroxyl groups on the surface of Fe3O4@SiO2 nanoparticles. XPS measurement and elemental analysis of MNPs-QN (see Supporting information) further confirmed the immobilization of quinine on the Fe3O4@SiO2 support. By the elemental analysis, the average loading of quinine was 0.5 mmol/g. Also, SEM measurements revealed the average granular particle size of MNPs-QN was 100–130 nm (Fig. S1 in Supporting information).

|
Download:
|
| Fig. 1. FT-IR spectra of the support, QN-immobilized Fe3O4@SiO2 (Ⅱ-QN), and the catalyst MNPs-QN. | |
The prepared MNPs-QN could be well dispersed in organic solvents like toluene to form a quasi-homogeneous suspension; on the other hand, MNPs-QN could be easily collected with the aid of an external magnet (Fig. S2 in the Supporting information). These properties allowed MNPs-QN to be used as a magnetically recoverable organocatalyst.
We then evaluated the catalytic efficiency and stereoselectivity of the prepared MNPs-QN and MNPs-QD as chiral organocatalysts. The Michael addition reactions of various carbon nucleophiles provide a powerful toolbox for the carbon-carbon bond formation in organic synthesis [32]. Also, both quinine and quinidine could efficiently catalyze highly enantioselective Michael addition reaction of 1, 3-dicarbonyls and maleimides [33]. Accordingly we chose this reaction to evaluate MNPs-QN and MNPs-QD. The preferable reaction conditions for MNPs-QN-catalyzed asymmetric Michael addition reaction were first surveyed with 1, 3-dicarbonyl compound 1a and maleimide 2a used as the model substrates (Table 1). Under the typical conditions and with the catalyst MNPs-QN (200 mg, 0.1 mmol) employed, the model reaction of cyclic β-ketoester 1a (1.5 mmol) and maleimide 2a (1.0 mmol) was respectively run in a series of common solvents for 24 h (Table 1, entries 1–7). The model reactions afforded the Michael addition product 3a in moderate to good yields with varied stereoselectivities. Polar acetonitrile and protic methanol were not suitable for this reaction with respect to stereoselectivity (entries 3 and 6); toluene was the preferable solvent, affording good yield and enantioselectivity (entry 7). For the purpose of comparison, the uncapped Ⅱ-QN was also tested as catalyst, giving inferior stereoselectivity (entry 8). The increase of the catalyst loading to 0.2 mmol and elongation of the reaction time to 48 h led to an improved yield and slightly better stereoselectivity (entry 9). Run at lower temperatures (0 ℃ and −30 ℃), the model reaction gave better stereoselectivities but slightly lower yields (entries 10 and 11). At 0 ℃, another catalyst MNPs-QD (0.2 mmol) was used instead of MNPs-QN, giving comparable yield and stereoselectivity. As expected, the opposite enantiomer of the major diastereomer 3a was obtained as the major product (Table 1, entry 12). Finally, the homogeneous quinine-catalyzed model reaction was also conducted, giving the same product 3a in comparable yield and enantioselectivity (entry 13). This result implies that the quinine-immobilized catalyst MNPs-QN behaves in the Michael reaction similarly to the homogeneous catalyst quinine. Considering both yield and stereoselectivity, the reaction conditions for entry 10 were chosen as the optimal.
|
|
Table 1 Screening of the reaction conditions of the asymmetric Michael reaction of 1a and 2a.a |

Under the optimal conditions, the scope of the asymmetric Michael addition reaction of 1, 3-dicarbonyls 1 and maleimides 2 was further explored (Table 2). The general procedure for the asymmetric Michael addition reaction of 1, 3-dicarbonyls 1 and maleimides 2: To a stirred mixture of maleimide 2 (1.0 mmol), supported catalyst MNPs-QN (400 mg, 0.2 mmol) in toluene (2.0 mL) was added 1, 3-dicarbonyl compound 1 (1.5 mmol). The resulting mixture was stirred under nitrogen and at 0 ℃ for 24–48 h. The solution was collected by decantation with the aid of a magnet and the remaining catalyst was washed with toluene (total 3.0 mL). The combined solution was concentrated under reduced pressure and the residue was subjected to flash column isolation to obtain the crude product 3 as an isomeric mixture, which was further purified by the second column chromatography on silica gel using petroleum ether (60–90 ℃)/ethyl acetate (5:1) as eluent to afford diastereomerically pure 3. The characterization data and NMR spectra (1H and 13C) of 3 are provided in Supporting information.
|
|
Table 2 Scope of the asymmetric Michael reaction of 1, 3-dicarbonyls 1 and maleimides 2. |

{kind=link}
{kind=link}
With the cyclic β-ketoester 1a employed as the representative substrate, a series of maleimides 2 bearing varied substituents at nitrogen atom were examined (Table 2, entries 1–7). Unsubstituted maleimide 2b gave the corresponding product 3b in moderate yield and modest enantioselectivity (for the major diastereomer) but in poor diastereoselectivity (entry 2). Alkyl-substituted maleimides 2c–2e gave moderate yields and moderate to excellent levels of both enantio-and diastereoselectivities (entries 3–5). Phenyl-substituted maleimide 2ffurnished the corresponding product 3f in good yield and enantioselectivity but in low diastereoselectivity (entry 6). Benzyl-substituted maleimide 2g was as effective as 2a, giving the corresponding product 3g in excellent yield and high stereoselectivity (entry 7). With maleimide 2a used as a reactant, a group of selected 1, 3-dicarbonyls 1b–1e were further investigated (entries 8–11). Cyclic β-ketoesters (1b and 1c) and 1, 3-diketone 1d readily afforded their corresponding products in high yields and stereoselectivities (entries 8–10). On the contrary, acyclic β-ketoester 1e effectively produced its addition product 3k in good yield and enantioselectivity but in poor diastereoselectivity (entry 11). Interestingly, the isolated product 3k as a diastereomeric mixture gradually converted into a diastereomerically pure product after being stored at r.t. for one week. Presumably, this conversion proceeds through a spontaneous keto-enol tautomerization mechanism. Except for 3k, the stereochemistry of products 3 was determined by analogy with the reported compounds [33].
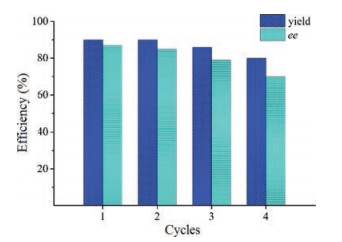
The reusability of the catalyst MNPs-QN was also tested through the model reaction of 1a and 2a run at r.t. for 48 h. After separation from the reaction mixture by decantation with the aid of an external magnet, the recovered catalyst MNPs-QN was washed 3 times with dry toluene and was directly used in the next batch of reaction. As Fig. 2 indicates, the catalyst suffered appreciable loss of both efficiency and stereoselectivity at its fourth use (Table S1 in Supporting information). After its third use, the recycled catalyst MNPs-QN was measured by IR spectroscopy. The IR spectrum of the recycled catalyst (Fig. 1, curve d) clearly indicated that the characteristic signals of the active quinine moiety remained intact. However, compared to the IR spectrum of the catalyst MNPs-QN (Fig. 1, curve c), the spectrum of the recycled catalyst obviously showed the broad signal of hydroxyl groups at about 3400 cm−1 in a low intensity. This result implies that the re-generation of free hydroxyl groups on the surface of the catalyst support may be responsible for the loss of the recycled catalyst's efficiency and stereoselectivity. The lower stereoselectivity of the uncapped Ⅱ-QN to some extent corroborates this hypothesis (Table 1, entry 8).

|
Download:
|
| Fig. 2. The reusability of the catalyst MNPs-QN. | |
{kind=link}
In conclusion, the magnetic nanoparticles supported cinchona alkaloids quinine and quinidine have been successfully synthesized for the first time as magnetically recoverable chiral organocatalysts. These prepared catalysts can efficiently catalyze asymmetric Michael addition reaction of 1, 3-dicarbonyls and maleimides in up to 93% ee. The reusability of the immobilized quinine catalyst was also evaluated and the catalyst can be consecutively used for up to 3 times without significant loss of efficiency. For the multiply recycled catalyst, the loss of efficiency and stereoselectivity may be attributed to the re-generation of the hydroxyl groups on the surface of the catalyst support. Further modifications on these catalysts and their applications in other organocatalytic asymmetric reactions are currently under investigation in our laboratories.
AcknowledgmentFinancial support from the National Natural Science Foundation of China (No. 21472096) is gratefully acknowledged.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.06.022.
| [1] |
K. Kacprzak, J. Gawronski, Synthesis(2001), 961-998. |
| [2] |
E.M.O. Yeboah, S.O. Yeboah, G.S. Singh, Tetrahedron 67(2011) 1725-1762. DOI:10.1016/j.tet.2010.12.050 |
| [3] |
J.D. Duan, P.F. Li, Catal. Sci. Technol. 4(2014) 311-320. DOI:10.1039/C3CY00739A |
| [4] |
L. Jiang, C. Y.-Chen, Catal. Sci. Technol. 1(2011) 354-365. DOI:10.1039/c0cy00096e |
| [5] |
P.F. Zheng, Q. Ouyang, S.L. Niu, et al., J. Am. Chem. Soc. 137(2015) 9390-9399. DOI:10.1021/jacs.5b04792 |
| [6] |
G. Zhan, M.L. Shi, Q. He, et al., Angew. Chem. Int. Ed. 55(2016) 2147-2151. DOI:10.1002/anie.201510825 |
| [7] |
M. Montesinos-Magraner, C. Vila, R. Canton, et al., Angew. Chem. Int. Ed. 54(2015) 6320-6324. DOI:10.1002/anie.201501273 |
| [8] |
F. Xue, X. Bao, L. Zou, J. Qu, B. Wang, Adv. Synth. Catal. 358(2016) 3971-3976. DOI:10.1002/adsc.201601070 |
| [9] |
S. Itsuno, M.M. Hassan, RSC Adv. 4(2014) 52023-52043. DOI:10.1039/C4RA09561H |
| [10] |
W.S. Zhao, Y.L. Zhang, C.K. Qu, et al., Catal. Lett. 144(2014) 1681-1688. DOI:10.1007/s10562-014-1322-5 |
| [11] |
C.B. Li, X.M. Shu, L. Li, et al., Chem. Asian J. 11(2016) 2072-2077. DOI:10.1002/asia.v11.14 |
| [12] |
I. Billault, R. Launez, M.C. Scherrmann, RSC Adv. 5(2015) 29386-29390. DOI:10.1039/C5RA02292D |
| [13] |
X.M. Xu, T.Y. Cheng, X.C. Liu, et al., ACS Catal. 4(2014) 2137-2142. DOI:10.1021/cs5002459 |
| [14] |
D. Cancogni, A. Mandoli, R.P. Jumde, D. Pini, Eur. J. Org. Chem.(2012), 1336-1345. |
| [15] |
Y.Y. Qin, G.X. Yang, L.L. Yang, J. Li, Y.C. Cui, Catal. Lett. 141(2011) 481-488. DOI:10.1007/s10562-010-0509-7 |
| [16] |
R. Porta, M. Benaglia, F. Coccia, F. Cozzi, A. Puglisi, Adv. Synth. Catal. 357(2015) 377-383. DOI:10.1002/adsc.201400821 |
| [17] |
J. Izquierdo, C. Ayats, A.H. Henselera, M.A. Pericas, Org. Biomol. Chem. 13(2015) 4204-4209. DOI:10.1039/C5OB00325C |
| [18] |
J.Q. Zhou, J.W. Wan, X.B. Ma, W. Wang, Org. Biomol. Chem. 10(2012) 4179-4185. DOI:10.1039/c2ob25106j |
| [19] |
G. Kardos, T. Soos, Eur. J. Org. Chem.(2013), 4490-4494. |
| [20] |
R.P. Jumde, A.D. Pietro, A. Manariti, A. Mandoli, Chem. Asian J. 10(2015) 397-404. DOI:10.1002/asia.v10.2 |
| [21] |
R.P. Jumde, A. Mandoli, F. De Lorenzi, D. Pini, P. Salvadori, Adv. Synth. Catal. 352(2010) 1434-1440. DOI:10.1002/adsc.v352:9 |
| [22] |
R. Mrowczynski, A. Nanb, J. Liebscher, RSC Adv. 4(2014) 5927-5952. DOI:10.1039/c3ra46984k |
| [23] |
R. Dalpozzo, Green Chem. 17(2015) 3671-3686. DOI:10.1039/C5GC00386E |
| [24] |
V. Angamuthu, D.F. Tai, Appl. Catal. A:Gen. 506(2015) 254-260. DOI:10.1016/j.apcata.2015.09.006 |
| [25] |
R.B.N. Baiga, M.N. Nadagoudab, R.S. Varma, Coord. Chem. Rev. 287(2015) 137-156. DOI:10.1016/j.ccr.2014.12.017 |
| [26] |
O. Gleeson, G.L. Davies, A. Peschiulli, et al., Org. Biomol. Chem. 9(2011) 7929-7940. DOI:10.1039/c1ob06110k |
| [27] |
X.X. Jiang, H. Zhu, X.M. Shi, et al., Adv. Synth. Catal. 355(2013) 308-314. |
| [28] |
J.W. Wan, L. Ding, T. Wu, X.B. Ma, Q. Tang, RSC Adv. 4(2014) 38323-38333. DOI:10.1039/C4RA04720F |
| [29] |
K. Rad-Moghadam, N. Dehghan, J. Mol. Catal. A:Chem. 392(2014) 97-104. DOI:10.1016/j.molcata.2014.05.005 |
| [30] |
Y. Lu, Y.D. Yin, B.T. Mayers, Y.N. Xia, Nano Lett. 2(2002) 183-186. DOI:10.1021/nl015681q |
| [31] |
A.K. Tucker-Schwartz, R.A. Farrell, R.L. Garrell, J. Am. Chem. Soc. 133(2011) 11026-11029. DOI:10.1021/ja202292q |
| [32] |
P. Pelmutter, Conjugate Addition Reactions in Organic Synthesis, Pergamon, Oxford, 1992.
|
| [33] |
G. Bartoli, M. Bosco, A. Carlone, et al., Angew. Chem. 118(2006) 5088-5092. DOI:10.1002/(ISSN)1521-3757 |