 2018, Vol. 29
2018, Vol. 29 



Quaternary coptisine (1, Fig. 1) is a typical natural quaternary protoberberine alkaloid (QPA). It was obtained previously from several Coptis species of the Ranunculaceae family and Corydalis species of the Papaveraceae family [1, 2]. The existence of two aromatic methylenedioxy groups at C-2/3 and C-9/10 positions, respectively, on the numbering system of QPA is its distinctive structural features compared with other natural QPAs. The 7, 8-imine salt functional group is one of its most active sites for chemical reaction. Quaternary coptisine was reported to possess certain biological and pharmacological activities worthy of inquiring for drug development, such as the properties of attenuating obesity-related inflammation [3], anti-alzheimer's disease [4], anti-hypercholesterolaema [5], anti-osteosarcoma [6], anti-fungus [7], cardioprotection [8, 9], and anti-proliferation of vascular smooth muscle cells [10, 11], among others [12-14]. But, by and large, extensive investigation into the medicinal chemistry of quaternary coptisine is relative scarce to date [15-17], especially compared with another well-known natural QPA, quaternary berberine. In view of this situation, certain structural modifications on quaternary coptisine and pharmacological investigations with the synthesized compounds were conducted recently in our laboratory. Several coptisine analogues were proved to be potential candidates for developing new drugs, such as anti-Ulcerative colitis (anti-UC) new drug [15-17].

|
Download:
|
| Fig. 1. Structure of 1. | |
Over the past several years, the selenoprotein thioredoxin reductases (TrxRs) as a potential target for cancer treatment attracted the attention of pharmacologists and medicinal chemists. The Michael acceptor of α, β-unsaturated carbonyl as a key structural feature was reported to be capable of improving the antitumor property of explored substrates and cancer chemotherapeutic drugs via a mechanism of targeting TrxRs inhibitively [18-26]. In our ongoing studies into the medicinal chemistry of quaternary coptisine, the advantage of 7, 8-imine salt functional group as chemically more active site than other structural moieties was again taken to carry out the structural modification to introduce an α, β-unsaturated ketone moiety, i.e., 1-acylethene-1-yl, at C-8 position. Eighteen target quaternary ammoniums were synthesized and different kinds of acyls were involved. In the course of synthesizing the target compounds, a methyl was inevitably introduced to C-13 when the Michael acceptor of α, β-unsaturated ketone moiety was built. Thus, the synthesized target compounds were actually quaternary 8-(1-acylethene-1-yl)-13-methylcoptisine chlorides. This paper describes the design, synthesis, and structural identification of target compounds. It also describes the pharmacological studies on the synthesized compounds, including the screening of the growth inhibitory activity against three human cancer cell lines, the effect on the viability of normal intestinal epithelial cell-6 (IEC-6) in vitro, the evaluation of efficacy by IC50 values as an indicator, and the structure-activity relationship (SAR) analysis of the growth inhibitory activity.
Under the conditions of building the α, β-unsaturated ketone moiety at C-8 via reactions of nucleophilic addition, aldol condensation, and dehydration of alcohol using methyl ketones and formaldehyde as reagents, a methyl was inevitably introduced at C-13 of the substrate. The synthesis of the desired quaternary 8-(1-acylethene-1-yl)-13-methylcoptisine chlorides as target compounds were modeled after our earlier study via the same threestep sequences as that indicated by Scheme 1. For a systematic goal to screen the growth inhibitory activity against the target cancer cell lines and to evaluate the SAR, the considered acyl groups involved n-alkanoyls, branched chain alkanoyls, cycloalkylcarbonyls, and aroyls. The n-alkanoyls involved those containing two to ten carbon atoms (a-i); the branched chain alkanoyls involved tert-butylcarbonyl (j) and iso-propylcarbonyl (k); the cycloalkylcarbonyls involved cyclopropylcarbonyl (l) and cyclohexylcarbonyl (m); and the aroyls involved 4-methoxybenzoyl (n), 4-isobutylbenzoyl (o), benzoyl (p), 2, 3-dihydrobenzo[b][1, 4]dioxin-6-ylcarbonyl (q), and naphthalen-1-yl-carbonyl (r). As pointed out in our previous study, in addition to the purification of (±)-8-acetonyldihydrocoptisine [(±)-2a] as one of the intermediates, which was achieved by recrystallization from acetone solvent, all the other key intermediates of (±)-8-acylmethyl-substituted dihydrocoptisines [(±)-2b-r] were directly applied in the next reaction step to conduct the syntheses of target compounds without any processing endeavor because there were no facile approach to separate and purify those intermediates. The experimental results affirmed the feasibility of this strategy. And, in addition to the reported modest yields between 22.2% and 71.5% from 1 for the synthesis of compounds 3a-d, 3j-l, 3n, and 3o, the new synthesized congeners 3e-i, 3m, and 3p-r were obtained at yields between 17.8% and 71.5% from 1. The structures of all the synthesized compounds were identified by the combination of the 1H NMR and 13C NMR data and ESIMS data (see Supporting information). In addition, some other chemical modifications of 1 were conducted in this study, including the reported inactive quaternary 13-methylcoptisine chloride (4a), which was considered as a pseudosubstrate for the synthesized compounds according to its structural features, and active quaternary 13-n-undecylcoptisine chloride (4l) and quaternary 13-n-dodecylcoptisine chloride (4m) [17], both of which were used as reference compounds and positive control to assess the effect of the α, β-unsaturated ketone moiety introduced into quaternary coptisine on the growth inhibitory activity against human cancer cell lines.

|
Download:
|
| Scheme 1. Reagents and conditions for syntheses of (±)-2a-r and 3a-r. (a) Methyl ketone, 5 mol/L NaOH, 60 ℃; (b) HCHO, THF, CH3COOH, reflux; (c) 2 mol/L HCl, CH3OH, r.t. | |
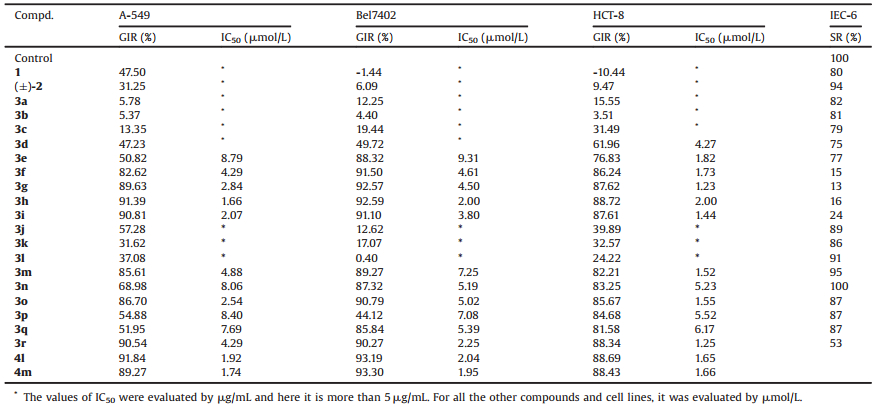
In order to evaluate the effect of the introduced α, β-unsaturated ketone moieties on the growth inhibitory activity of the target compounds against human cancer cell lines and to clarify the SAR, all the synthesized compounds 3a-r were investigated for their activity in vitro using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-2H-tetrazolium bromide (MTT) assay. Quaternary coptisine (1), quaternary 13-methylcoptisine chloride (4a), quaternary 13-n-undecylcoptisine chloride (4l), and quaternary 13-n-dodecylcoptisine chloride (4m) were screened in the same batches of experiments. The explored human cancer cell lines included human lung adenocarcinoma (A-549), human hepatoma (Bel7402), and human colorectal cancer (HCT-8) cell lines. On evaluation of these compounds, all target cancer cells were treated successively using every tested compound for 96 h, respectively. The MTT reduction assay procedure was modeled after our earlier study [17]. Results of means of three replicates presented in Table 1 were expressed as the concentration for samples to inhibit the cell growth by 50%, i.e., the IC50 values, when the growth inhibition rate (GIR) of tested compounds was more than the value of 50%. Among the tested compounds, the active quaternary 8-(1-acylethene-1-yl)-13-methylcoptisine chlorides were defined by the IC50 values in the range of 1.23–9.31 μmol/L. Several compounds were found to show the IC50 values at the same level as those of the reference compounds, 4l and 4m, in the present study. On this benchmark, the active compounds covered those with acyls being n-alkanoyls containing six to ten carbon atoms (3e-i) or cyclohexylcarbonyl (3m) or various aroyls (3n-r). Compound 3d also showed activity against HCT-8 cell line. The inactive compounds included those possessing n-alkanoyls containing two to four carbon atoms (3a-c) or tert-butylcarbonyl (3j) or iso-propylcarbonyl (3k) or cyclopropylcarbonyl (3l). From the viewpoint of SAR, some features involving the impactof structureon target activitycan be explicitly pointed out. The introduction of the 1-acylethene-1-yl groups of long-chain and more lipophilic n-alkanoyls into C-8 position of lead compound (or the reported inactive 4a) played a worthy role for improving the growth inhibitory activity against the three human cancer cell lines. Especially, the growth inhibitory activity of the synthesized quaternary 8-(1-n-alkanoylethene-1-yl)-13-methylcoptisine chlorides against all the three tested cell lines were enhanced as the length of the aliphatic chain of the n-alkanoyl was elongated if overriding the deviation of individual data (Fig. 2). It was observed that quaternary 8-(1-capryloylethene-1-yl)-13-methylcoptisine chloride (3g), quaternary 8-(1-nonanoylethene-1-yl)-13-methylcoptisine chloride (3h), and quaternary 8-(1-decanoylethene-1-yl)-13-methylcoptisine chloride (3i) showed the most significant, and almost the same level of, activity among the investigated 8-(1-n-alkanoylethene-1-yl)-13-methylcoptisine chlorides by their IC50 values ranging from 1.23 μmol/L to 4.50 μmol/L (Table 1). The two reference compounds, 4l and 4m, displayed IC50 values of 1.92, 2.04, and 1.65 μmol/L and 1.74, 1.95, and 1.66 μmol/L, respectively, against A-549, Bel7402, and HCT-8 cell lines in the control test. The synthesized quaternary 8-(1-n-alkanoylethene-1-yl)-13-methylcoptisine chlorides with the n-alkanoyls possessing six (3e) or seven (3f) carbons also displayed moderate or significant growth inhibitory activity against the tested cancer cell lines. The IC50 values of compounds 3e and 3f were 8.79, 9.31, and 1.82 μmol/L and 4.29, 4.61, and 1.73 μmol/L, respectively, against A-549, Bel7402, and HCT-8 cell lines. This conclusion about the SAR of the synthesized compounds containing n-alkyls was very compatible with a similar investigation on related QPAs reported in a previous study [27]. All the synthesized 8-(1-aroylethene-1-yl)-13-methylcoptisine chlorides (3n-r), as well as 8-(1-cyclohexylcarbonylethene-1-yl)-13-methylcoptisine chloride (3m), also displayed significant target activityagainst the three tested human cancer cell lines when evaluated by their IC50 values rang of 1.25– 8.40 μmol/L, but no SAR was observed for the tested aroyls themselves (Fig. 3). This investigation cannot be counted on to draw a final conclusion about the SAR of various aroyls, say, what would the influence of electron-withdrawing groups and electrondonating groups linked to the aromatic moieties be on the activity, because of the relatively small number of tested aroyls involved in this study. But, obviously, phenyl, 4-methoxyphenyl, 4-isobutylphenyl, 2, 3-dihydrobenzo[b][1, 4]dioxin-6-yl, and naphthalen-1-yl, among others, possessed the relatively similar lipophilicity. Therefore, the findings about SAR of lipophilicity were produced. In contrast with the n-alkanoyls, aroyls, and cyclohexylcarbonyl in the 1-acylethene-1-yl of active compounds, the acyls containing relatively more polar groups, including n-alkanoyls of possessing two to four carbon atoms (3a-c), branched chain alkanoyls of possessing tert-butyl (3j) or iso-propyl (3k), and cyclopropylcarbonyl (3l), displayed no effect on improving the growth inhibitory activity of target compounds against the tested human cancer cell lines. The observed worthy correlations between the introduction of 1-n-alkanoylethene-1-yls containing relatively more lipophilic long-chain alkyls at C-8 position of the substrate and the increased target activities and between the introduction of 1-aroylethene-1-yls at C-8 position and the increased target activities, as well as the obviously different situations between introducing 1-cyclopropylcarbonylethene-1-yl and 1-cyclohexylcarbonylethene-1-yl, would suggested that lipophilicity, for the most part, contributes to the tested activities as one of the key factors.
|
|
Table 1 Growth inhibitory activities of 1, (±)-2, 3a-r, 4l, and 4m against human cancer A-549, Bel7402, and HCT-8 cell lines and SRs on normal IEC-6 cell in vitro. |
{kind=link}
{kind=link}

|
Download:
|
| Fig. 2. Growth inhibitory activity and SARof compounds 3a-i against human cancer cell lines. The IC50 values of inactive compounds 3a-c for all the three tested cell lines and compound 3d for A-549 and Bel7402 were evaluated by μg/mL and here the values are more than 5 μg/mL. For all the other compounds and cell lines, it was evaluated by μmol/L. | |
{kind=link}

|
Download:
|
| Fig. 3. Growth inhibitory activity of compounds 3n-r (aroyls; a: 4-methoxybenzoyl; b: 4-isobutylbenzoyl; c: benzoyl; d: 2, 3-dihydrobenzo[b][1, 4]dioxin-6-ylcarbonyl; e: naphthalen-1-yl-carbonyl) against human cancer cell lines. | |
{kind=link}
In view of the reported clarification of Michael acceptor-centric pharmacophores that yield strong TrxR-inhibitory character correlated to antiproliferative activity [22-26, 28] and the achievement of our original intention to improve the growth inhibitory activities of the substrate against human cancer cell lines via introducing an α, β-unsaturated ketone moiety at C-8, targeting the cellular redox system might be a possible pathway for accounting for the observed growth inhibitory activity of the synthesized compounds.
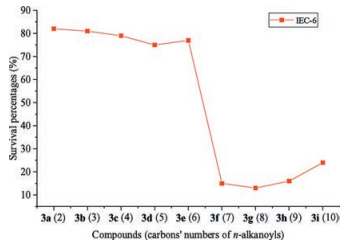
Another meaningful finding in the current investigation was that some aforementioned active compounds showed up selectivity for inhibiting the cell growth of target human cancer cell lines. It is well known that compounds with low cytotoxicity may be very interesting in exploring other biological activities, such as, the activities of anti-UC and anticancer, and so forth. Thus, considering the selected human cancer cell lines in this study, all the synthesized quaternary 8-(1-acylethene-1-yl)-13-methylcoptisine chlorides were investigated for their effect on the viability of normal IEC-6 cell in vitro using the MTT assay at a constant concentration of 10 μmol/L with blank control group. The result showed that compounds 3f-i, which are the compounds with nonpolar n-alkanoyls possessing seven or more carbons, displayed significant cytotoxicity on normal IEC-6 cell with survival rates (SRs) of 15%, 13%, 16%, and 24%, respectively, and compound 3r displayed a low cytotoxicity on normal IEC-6 cell with SR of 53% when co-incubated with the tested normal cell for 24 h (see Supporting information). None of the other compounds displayed obvious cytotoxicity on normal IEC-6 cell when co-incubated. The SRs of the noncytotoxic compounds ranged from 75% to 100% (Table 1). The result after 3 days of co-incubation was the same as that after 24 h (data not shown). Thus, a possibility emerged that, although the elongation of the aliphatic chain of the n-alkanoyls improved the growth inhibitory activity of target compounds against the tested cancer cell lines, it also increased the inhibitory action on the cell growth of normal IEC-6 cell. As soon as the aliphatic chain grew beyond five carbon atoms, i.e., the n-alkanoyls possessed seven or more carbons, the target compounds displayed cytotoxicity on normal IEC-6 cell (Fig. 4).

|
Download:
|
| Fig. 4. Results of toxicity test of compounds 3a-i on IEC-6 cell determined by MTT assay in vitro. | |
{kind=link}
The findings of this study suggested that chemical modifications of natural quaternary coptisine by introducing certain 1-acylethene-1-yl groups into C-8 position, combining the simultaneous introduction of a methyl group at C-13, can remarkably improve the growth inhibitory activity against the target human cancer cell lines. The activity of target compounds was significantly graced as the oil/water partition coefficients of the acyls in the 1-acylethene-1-yl group increased. Thus, this reported structural modification is likely to have made the compounds more lipophilic or to possess a more suitable dissociation degree in body fluid in nature, which in turn may have made them easier in crossing over the lipid phase-water phase-lipid phase biomembranes to enter body fluids for absorption and distribution. And thus, the chemical modification of this study is more effective for improving the activity of target compounds in inhibiting the growth of tested human cancer cells based on the comprehensive function of the structure of lead compound. On the other hand, the findings that some compounds showed nearly no cytotoxicity when coincubated with normal IEC-6 cells was also interesting. It suggested that these active compounds showed up not just inhibitory activity for the cell growth of tested human cancer cell lines, but also suitability for screening other downstream biological activities with normal IEC-6 cell models, i.e., the selective growth inhibitory activity against human cancer cell lines and normal IEC-6 cell was found. In addition, according to the reported data, targeting the cellular redox system might be a possible pathway for accounting for the observed growth inhibitory activity of the synthesized compounds against human cancer cell lines. To sum up, a new class of promising compounds for developing anti-cancer drugs against human lung cancer or liver cancer or colorectal cancer, or for studying other bioactivities, could be proposed to prepare from quaternary coptisine as starting substance. The hydrogen atom at C-8 of the lead compound could be substituted by an α, β-unsaturated ketone group of 1-acylethene-1-yl of long-chain n-alkanoyl possessing six carbons or of aroyls or cyclohexylcarbonyl when the hydrogen atom at C-13 was also substituted by a methyl. This result may also guide future rational design efforts in medicinal chemistry of quaternary coptisine.
AcknowledgmentsThis work was supported by grants from the National Natural Science Foundation of China (No. 81373269), CAMS Innovation Fund for Medical Sciences (No. 2016-12M-1-010), and National Science and Technology Project of China (No. 2017ZX09305008-002).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.08.026.
| [1] |
W. Wang, Q.W. Zhang, W.C. Ye, et al., Chin. J. Nat. Med. 5(2007) 348-350. |
| [2] |
L. Grycová, J. Dostál, R. Marek, Phytochemistry 68(2007) 150-175. DOI:10.1016/j.phytochem.2006.10.004 |
| [3] |
Z.Y. Zou, Y.R. Hu, H. Ma, et al., Fitoterapia 105(2015) 139-146. DOI:10.1016/j.fitote.2015.06.005 |
| [4] |
D. Yu, B.B. Tao, Y.Y. Yang, et al., J. Alzheimers Dis. 43(2015) 291-302. |
| [5] |
K. He, X. Ye, H. Wu, et al., Lipids 50(2015) 185-194. DOI:10.1007/s11745-014-3983-7 |
| [6] |
D. Yu, S. Fu, Z. Cao, et al., Toxicol. Lett. 226(2014) 328-336. DOI:10.1016/j.toxlet.2014.02.021 |
| [7] |
W.J. Kong, Y.L. Zhao, X.H. Xiao, et al., J. Appl. Microbiol. 107(2009) 1072-1080. DOI:10.1111/j.1365-2672.2009.04292.x |
| [8] |
J. Guo, S.B. Wang, T.Y. Yuan, et al., Atherosclerosis 231(2013) 384-391. |
| [9] |
L.L. Gong, L.H. Fang, S.B. Wang, et al., Atherosclerosis 222(2012) 50-58. DOI:10.1016/j.atherosclerosis.2012.01.046 |
| [10] |
H. Tanabe, H. Suzuki, H. Mizukami, et al., Biochem. Pharmacol. 70(2005) 1176-1184. DOI:10.1016/j.bcp.2005.07.010 |
| [11] |
H. Suzuki, H. Tanabe, H. Mizukami, et al., J. Nat. Prod. 74(2011) 634-638. DOI:10.1021/np100645d |
| [12] |
H. Suzuki, H. Tanabe, H. Mizukami, et al., Biol. Pharm. Bull. 33(2010) 677-682. DOI:10.1248/bpb.33.677 |
| [13] |
Z.H. Zhang, A.J. Deng, J.Q. Yu, et al., China J. Chin. Mater. Med. 38(2013) 2750-2754. |
| [14] |
J.W. Lee, A. Iwahashi, S. Hasegawa, et al., J. Nat. Med. 66(2012) 8-16. DOI:10.1007/s11418-011-0537-7 |
| [15] |
M. Xie, H.J. Zhang, A.J. Deng, et al., J. Nat. Prod. 79(2016) 775-783. DOI:10.1021/acs.jnatprod.5b00807 |
| [16] |
Z.H. Zhang, H.J. Zhang, A.J. Deng, et al., J. Med. Chem. 58(2015) 7557-7571. DOI:10.1021/acs.jmedchem.5b00964 |
| [17] |
Z.H. Zhang, A.J. Deng, L.Q. Wu, et al., Eur. J. Med. Chem. 86(2014) 542-549. DOI:10.1016/j.ejmech.2014.09.006 |
| [18] |
L.R. Huang, H. Luo, X.S. Yang, et al., Med. Chem. Res. 23(2014) 4631-4641. DOI:10.1007/s00044-014-1031-z |
| [19] |
I. Karpavičienė, G. Valiulienė, V. Raškevičius, et al., Eur. J. Med. Chem. 98(2015) 930-48. |
| [20] |
S. Xu, S. Luo, H. Yao, et al., J. Med. Chem. 59(2016) 5022-5034. DOI:10.1021/acs.jmedchem.6b00408 |
| [21] |
L.D. Liu, S.L. Liu, Z.X. Liu, et al., J. Mol. Struct. 1112(2016) 1-8. DOI:10.1016/j.molstruc.2016.02.025 |
| [22] |
B. Zhang, D. Duan, C. Ge, et al., J. Med. Chem. 58(2015) 1795-1805. DOI:10.1021/jm5016507 |
| [23] |
D. Duan, J. Zhang, J. Yao, et al., J. Biol. Chem. 291(2016) 10021-10031. DOI:10.1074/jbc.M115.700591 |
| [24] |
J. Zhang, Y. Li, D. Duan, et al., Biochem. Pharmacol. 102(2016) 34-44. DOI:10.1016/j.bcp.2015.12.004 |
| [25] |
J. Zhang, J. Yao, S. Peng, et al., Biochim. Biophys. Acta 1863(2017) 129-138. DOI:10.1016/j.bbadis.2016.10.019 |
| [26] |
J. Fang, A. Holmgren, J. Am. Chem. Soc. 128(2006) 1879-1885. DOI:10.1021/ja057358l |
| [27] |
K. Iwasa, M. Moriyasu, T. Yamori, et al., J. Nat. Prod. 64(2001) 896-898. DOI:10.1021/np000554f |
| [28] |
F.F. Gan, K.K. Kaminska, H. Yang, et al., Antioxid. Redox Signal. 19(2013) 1149-1165. DOI:10.1089/ars.2012.4909 |