 2018, Vol. 29
2018, Vol. 29 



b Key Laboratory of Guizhou High Performance Computational Chemistry, Guizhou University, Guiyang 550025, China;
c Key Laboratory of Chemistry for Natural Products of Guizhou Province, Guiyang 550002, China
The construction of nanometer-scale devices such as molecular machines and switches from molecular components is of major interest in modern science and technology [1]. Mechanically interlocked molecules (MIMs) such as rotaxanes and shuttles have great potential as such molecular devices because the relative positions of their components can be induced to change by external chemical, electrochemical or photochemical stimuli [2]. Fluorescent signals which can be attributed to a distinct emission response due to changes in the physical and chemical properties in various microenvironments, have drawn much attention in recent years [3]. In particular, real-time and real-space analysis of a dynamic motion in appropriately designed MIMs systems is proving to be particularly popular [4].
Cucurbit[n]urils (Q[n]s or CB[n]s) [5] are a relatively new class of macrocyclic receptors and add to the growing number of macrocycles in the literature which already includes crown ethers, cryptands, cyclodextrins, and calixarenes. Q[n]s bear a rigid hydrophobic cavity and two identical carbonyl fringed portals. They are finding use in both supramolecular chemistry and materials science, which is a result of their superior molecular recognition properties in aqueous media [6, 7]. A recent study on the interactions of Q[n]s with fluorophore guest molecules revealed that Q[n]s can significantly modify the physicochemical properties of guest molecules upon complexation [8]. However, there are few reports in the literature on fluorescent rotaxane switches derived from cucurbiturils [9]. The major challenge in the construction of such switches is the lack of suitable water soluble fluorescent axle molecules. From a structural viewpoint, the rigid hydrophobic cavities and the polar portals available to cucurbiturils make them ideal host components for the target fluorescent switches.
In Q[n]-based MIMs, the vast majority of axle molecules have been derived from pyridinium and ammonium ions [10]. Surprisingly, despite several advantages available to bispyridinium ethylene derivatives versus viologen derivatives, interaction of the former group of guests with Q[n] molecules has been significantly less explored, particularly with respect to their fluorescence properties [11]. Recently, a number of bispyridinium ethylene-core based cationic derivatives have been combined with cucurbiturils and via the resulting host−guest chemistry have been employed by Zhang and co-workers for rationally adjusting multicolor emissions [12]. However, there are no studies on such guests employed as the axle to construct MIMs systems that can be monitored by fluorescent signals.
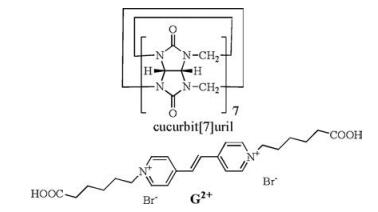
In the present study, we report the host-guest interaction between Q[7] and a 1, 2-di(4-pyridyl)ethylenyl derived dicationic fluorescent axle guest G2+ (Scheme 1), which gives rise to pseudorotaxanes that can be employed as a smart switch via the observation of distinct fluorescent signals in aqueous solution.

|
Download:
|
| Scheme 1. Chemical structures of Q[7] and G2+. | |
{kind=link}
As shown in Scheme 1, the hexanoic acid group was selected here as the end unit at the bispyridinium ethylene as this group not only increases the aqueous solubility of the guest but also reversibly switches between neutral and anionic states (depending on the pH of the media). For example, Kaifer and co-workers previously appended two identical carboxylic acids to the 4, 4-bipyridinium (viologen) core and found that the Q[7]-based pseudorotaxane behaved like a molecular shuttle when the two terminal-COOH units were protonated. By contrast, Q[7] simply located itself around the aromatic viologen unit at high pH [13].
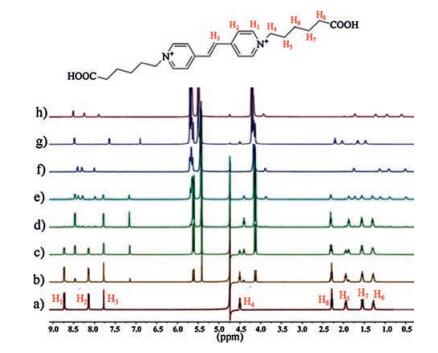
Fig. 1 shows the 1H NMR titration spectra of the G2+ guest in D2O recorded in the absence and presence of different concentrations of host Q[7]. Upon the gradual addition of the Q[7] host (below 1.0 equiv.) to a solution of G2+ at pH 2.0, the resonances corresponding to the protons on the bispyridinium ethylene-core were found to split into two sets of signals. This can be attributed to the chemical exchange rate between the free guest and the Q[7]-bound guest being slow on the NMR time scale. In the presence of 1.0 equiv of Q[7], the original aromatic proton signals associated with the bispyridinium ethylene disappeared. As shown in Fig. 1d, significant 1H NMR upfield shifts were noted for the protons H1, H2, H3, and H4, whereas no significant chemical shift changes for the protons on the aliphatic chain were observed. This result indicated that only the bispyridinium ethylene core of G2+ is encapsulated by the hydrophobic cavity of Q[7] rather than the whole G2+ guest. In other words, under acidic conditions, the 1:1 pseudorotaxane complex of Q[7] with G2+ in the present study resulting Q[7] host shuttles back and forth on the bispyridinium ethylene core. This host-guest behavior is very different from that reported for the Kaifer's system [13]. In that case, the Q[7] host shuttles back and forth between the two aliphatic ends of the viologen based guest. On the other hand, in 2011, Sindelar et al. successfully constructed a supramolecular shuttle based on the inclusion complex between Q[6] and bispyridinium ethylene [11]. However, their results suggested that the shuttling of Q[6] on the bispyridinium ethylene group is very slow. Our study here indicated that the Q[7] shuttle based on bispyridinium ethylene-core is fast than that observed for Q[6].

|
Download:
|
| Fig. 1. 1H NMR spectra (500 MHz, D2O, pD 2.0) of G2+ (2.0 mmol) in the absence of Q [7] (a) and in the presence of 0.25 equiv. Q[7] (b), 0.5 equiv. Q[7] (c), 1.0 equiv. Q[7] (d), 1.5 equiv. Q[7] (e), 2.0 equiv. Q[7] (f), addition of NaOD to the solution of f ([2] pseudorotaxane formed) (g), addition of DCl to the solution of g ([3] pseudorotaxane formed) (h). | |
{kind=link}
Further addition of the host Q[7] to the solution leads to the appearance of a new series of resonances. For example, in the presence of 2.0 equiv. of Q[7] (Fig. 1f), all of the protons on the aliphatic chain exhibit a large upfield shift. On the contrary, the signals in the spectra corresponding to the aromatic protons, especially protons H2 and H3 are shifted downfield compared to their original signals. These proton changes can be attributed to the presence of [3]pseudorotaxanes (2:1 complex of Q[7] with G2+). Interestingly, when the pH of the solution is raised to 10, it was found that the proton changes of G2+ were similar as the case of 1:1 pseudorotaxane complex of Q[7] with G2+ (Fig. 1g), whilst the [3]pseudorotaxanes can be recovered at lower pH values (Fig. 1h). This experiment clearly demonstrated that the pseudorotaxane modes of Q[7] with G2+ can be switched by the terminal carboxylates groups, which was previously observed in viologen-derived axle system. However, as mentioned by Kaifer et al, in the viologen system (1:1, host/guest), if the pH of the solution is raised enough to cause the deprotonation of the terminal carboxylates, the Q[7] host then locates itself around the aromatic viologen unit, residing on this central binding site thereby discontinuing the shuttling motions between the terminal aliphatic sites. In the present study, as illustrated in Scheme 2, we found that the Q[7]-based [2]pseudorotaxane behaves like a molecular shuttle on the bispyridinium ethylene core and can survive in both states presented by the carboxylates (i.e., protonation/deprotonation) and different temperature (Fig. S1 in Supporting information).

|
Download:
|
| Scheme 2. Schematic representation of the shuttling and presudorotaxane modes of the Q[7] wheel. | |
{kind=link}
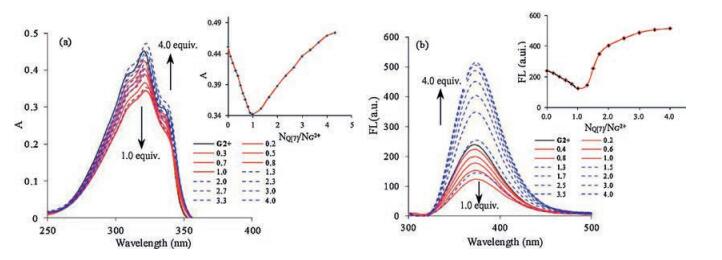
In an effort to gain more detailed information on the host-guest interactions between Q[7] and G2+, the differing pseudorotaxanes modes were evaluated by UV–vis absorption spectra and fluorescence spectra in aqueous solution at different pH values (Fig. S2 in Supporting information). As shown in Fig. 2a, the UV–vis absorption spectrum of free G2+ in aqueous solution (pH 2.0) features a maximum absorption peak at 320 nm. Following the addition of increasing concentrations of Q[7] to the solution of G2+, the intensity of the absorption maximum associated with G2+ gradually decreased until the concentration of Q[7] reached 1.0 equiv. However, it was noted that the maximum absorption intensity at 320 nm was gradually recovered and enhanced on further addition of host Q[7] to the solution. A plot of absorbance intensity at 320 nm versus n (Q[7]/G2+, mole ratio) clearly indicated two different binding models existed for the Q[7]/G2+ system.

|
Download:
|
| Fig. 2. (a) UV–vis spectra of G2+ (10.0 mmol/L) with increasing concentrations of Q[7] in aqueous solution (pH 2.0) at 298 K. Inset: curve of A vs. n (Q[7]/G2+) for the absorption peak at 320 nm. (b) Fluorescence spectra of G2+ (10.0 mmol/L) with increasing concentrations of Q[7] in aqueous solution (pH 2.0) at 298 K. λex = 321 nm. Inset: curve of fluorescence intensity vs. n (Q[7]/G2+) for the emission peak at 373 nm. | |
{kind=link}
Interestingly, similar fluorescent response behavior was observed for Q[7] with G2+ under the same analytical conditions. As illustrated in Fig. 2b, guest G2+ itself exhibited a characteristic maximum emission band at 373 nm with an excitation wavelength at 321 nm. No shift in the maximum of the emissions of G2+ was observed upon the addition of the host Q[7] to the solution, but the fluorescence intensity of the guest markedly decreased when the concentration of Q[7] was raised from 0 to 1.0 equiv, and then significantly increased when the concentration of Q[7] exceeded 1.0 equiv. A plot of emission intensity at 373 nm versus n (Q[7]/G2+, mole ratio) provided unequivocal proof of the 1:1 and 2:1 binding models for the Q[7]/G2+ system. The corresponding formation constants were determined to be K1:1 = (2.5 ± 0.23) × 105 L/mol, and K2:1 = (7.02 ± 0.15) × 106 L/mol, respectively. Therefore, based on these observations, the two different host-guest interactions of Q[7] with G2+ can be distinguished by the fluorescent signal. In other words, the [2]pseudorotaxane is fluorescence "off" and the [3]pseudorotaxane is fluorescence "on".
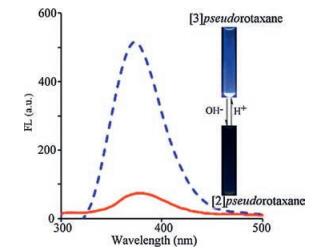
As revealed in Fig. 1, 1H NMR spectroscopic results suggested that the pseudorotaxanes modes of Q[7] with G2+ can be switched by the terminal carboxylates groups, because of the strong electrostatic repulsions between the cavity opening of Q[7] and the anionic carboxylate at either end of the axis of the molecule. To further evaluate whether such molecular motions can be monitored by the fluorescence signal, an estimation of the fluorescence response of Q[7]/G2+ system was conducted at different pH values. For example, as shown in Fig. 3, at pH 2.0, the fluorescence of the Q[7]/G2+ system exhibited a clear enhancement, indicating that [3]pseudorotaxane was formed. When the pH of the solution was raised to 10.0, the related fluorescence emission intensity was significantly decreased, suggesting a motion transfer of Q[7]/G2+ system from [3]pseudorotaxane to [2]pseudorotaxane, in line with the fluorescence results presented in Fig. 2b. On further adjusting the pH value of the solution to the lower region, an enhancement in the fluorescence intensity could again be observed. These observations demonstrated that the different molecular motions between Q[7] and G2+ can be controlled and visually monitored.

|
Download:
|
| Fig. 3. pH switched pseudorotaxanes modes between Q[7] and G2+ with fluorescence signal. | |
{kind=link}
In cucurbiturils chemistry, in particular for Q[7], the formation of host-guest complexes might have restricted the rotational and vibrational motions of the aromatic fluorophore in the hydrophobic cavity. This restriction occurs via non-radiative, excited-state decay processes, and thus an enhancement in the emission of the fluorophores upon complexation generally can be observed. Unexpectedly therefore, it was surprised that a quenching fluorescence emission of G2+ occurred upon complexation with Q[7] in the procedure used for the formation of [2]pseudorotaxane in this work. In order to illustrate this phenomenon, the structures of G2+ with Q[7] were optimized by using density functional theory (DFT). As shown in Fig. 4, the energy-minimized structures of Q[7]/G2+ when considered alongside the 1H NMR spectra changes gives useful information. In the structure of [2]pseudorotaxane (Fig. 4a), the steric exclusion of Q[7] host to the pyridinium ethylene moiety on guest G2+ forces the styrene group to turn at an angle and twist the plane between the two pyridinium rings, which enables the G2+ to undergo an efficient formation of twisted intramolecular charge transfer (TICT)[14]. In other words, TICT is the predominant decay process of the excited states in the [2]pseudorotaxane structure due to the host-guest interaction, and fluorescence quenching occurred. In contrast, in the presence of a second Q[7] molecule (Fig. 4b), the modeling shows that the original host molecule is forced to abandon its inclusion of the pyridinium ethylene group in favor of the unoccupied alkyl chain. Obviously, the simultaneous inclusion of the alkyl chain and an adjacent pyridinium ethylene group is less stable here because of the unfavorable steric and electrostatic interactions between the hosts in these locations. In particular, the polar carbonyl-rimmed portals would result in significant repulsive dipole-dipole electrostatic interactions between two Q[7] hosts when placed over neighboring regions of the guest. As a result, each portal of the host is adjacent to its own site of positive change, maximizing the number of ion-dipole interactions between the guest and host, which results in the two pyridinium ethylene groups being located on a plane and thus resulting in strong suppression of the TICT of G2+. Accordingly, an enhancement fluorescent emission was achieved and clearly observed in the [3]pseudorotaxane system.

|
Download:
|
| Fig. 4. DFT-optimized structure of Q[7] with G2+ (a) [2]pseudorotaxane, and (b) [3] pseudorotaxane, respectively, at the B3LYP/6-311G (d, p) level of theory with Gaussian16 (hydrogen atoms are omitted for clarity). | |
{kind=link}
In summary, we have studied the detailed host-guest triggered pseudorotaxane modes between Q[7] and the bispyridinium ethylene-core derived guest G2+ by 1H NMR spectra and fluorescent signal changes. The proton shifts of G2+ induced by the host suggested that the Q[7]-based [2]pseudorotaxane behaves like a fast molecular shuttle on the bispyridinium ethylene axle when the guest is both protonation and deprotonation at the terminal carboxylates groups. Furthermore, the distinct fluorescent response behavior indicated that the bispyridinium ethylene moiety can not only behave as the axle component for the pseudorotaxane system, but also acts as an optical signaling unit during the host–guest complexation. We believe that the novel axle and photophysical properties of the bispyridinium ethylene will be of interest to the multidisciplinary fields of molecular machines and organic materials.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21302026, 21361006), "Chun-Hui" Fund of Chinese Ministry of Education (Nos. Z2016011, Z2015002), and Guizhou Province (Nos. 20165656, 20132150).
| [1] |
(a) V. Balzani, A. Credi, F. M. Raymo, J. F. Stoddart, Angew. Chem. Int. Ed. 112(2000) 3484-3530; (b) M. Gómez-López, J. A. Preece, J. F. Stoddart, Nanotechnology 7(1996) 183-192. |
| [2] |
(a) R. S. Forgan, J. P. Sauvage, J. F. Stoddart, Chem. Rev. 111(2011) 5434-5464; (b) D. H. Qu, H. Tian, Chem. Sci. 2(2011) 1011-1015; (c) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh, R. T. McBurney, Angew. Chem. Int. Ed. 50(2011) 9260-9327; (d) S. H. Li, Y. M. Zhang, Y. Liu, Chin. Sci. Bull. 61(2016) 3917-3923; (e) M. Xue, Y. Yang, X. D. Chi, X. Z. Yan, F. H. Huang, Chem. Rev. 115(2015) 7398-7501; (f) X. Wu, L. Gao, J. Z. Sun, X. Y. Hu, L. Y. Wang, Chin. Chem. Lett. 27(2016) 1655-1660; (g) H. Wang, Z. J. Zhang, H. Y. Zhang, Y. Liu, Chin. Chem. Lett. 24(2013) 563-567; (h) L. L. Hu, W. Xue, J. Yin, Chin. Chem. Lett. 27(2016) 155-158. |
| [3] |
(a) B. Valeur, Molecular Fluorescence: principles and Applications, Wiley-VCH, Weinheim, Germany, 2002; (b) V. Balzani, A. Credi, M. Venturi, Molecular Devices and Machines-concepts and Perspectives for the Nanoworld, Wiley-VCH, Weinheim Germany, 2008. |
| [4] |
(a) H. Wang, X. F. Ji, Z. T. Li, F. H. Huang, Adv. Mater. 29(2017) 1606117; (b) H. Zhang, J. Hu, D. H. Qu, Org. Lett. 14(2012) 2334-2337; (c) X. F. Ji, Y. Yao, J. Y. Li, X. Z. Yan, F. H. Huang, J. Am. Chem. Soc. 135(2013) 74-77; (d) T. T. Cao, X. Y. Yao, J. Zhang, Q. C. Wang, X. Ma, Chin. Chem. Lett. 26(2015) 867-871. |
| [5] |
(a) J. Kim, I. S. Jung, S. Y. Kim, et al., J. Am Chem. Soc. 122(2000) 540-541; (b) A. I. Day, A. P. Arnold, R. J. Blanch, B. Snushall, J. Org. Chem. 66(2001) 8094-8100. |
| [6] |
(a) S. J. Barrow, S. Kasera, M. J. Rowland, J. D. Barrio, O. A. Scherman, Chem. Rev. 115(2015) 12320-12406; (b) X. L. Ni, X. Xiao, H. Cong, et al., Acc. Chem. Res. 47(2014) 1386-1395. |
| [7] |
(a) W. Zhang, Y. M. Zhang, S. H. Li, et al., Angew. Chem. Int. Ed. 55(2016) 11452-11456; (b) M. H. Tootoonchi, G. Sharma, J. Calles, R. Prabhakar, A. E. Kaifer, Chem. Int. Ed. 55(2016) 11507-11511; (c) J. Tian, Z. Y. Xu, D. W. Zhang, et al., Nature Commun. 7(2016) 11580; (d) Q. Zhang, D. H. Qu, Q. C. Wang, H. Tian, Angew. Chem. Int. Ed. 127(2015) 16015-16019; (e) L. C. Smith, D. G. Leach, B. E. Blaylock, O. A. Ali, A. R. Urbach, J. Am. Chem. Soc. 137(2015) 3663-3669; (f) H. Li, Y. W. Yang, Chin. Chem. Lett. 24(2013) 545-552. |
| [8] |
G. Ghale, W.M. Nau, Acc. Chem. Res. 47(2014) 2150-2159. DOI:10.1021/ar500116d |
| [9] |
(a) X. L. Ni, S. Y. Chen, Z. Y. P. Yang, J. Am. Tao, Chem. Soc. 138(2016) 6177-6183; (b) S. K. Samanta, K. G. Brady, L. Isaacs, Chem. Commun. 53(2017) 2756-2759; (c) S. Q. Xu, X. Zhang, C. B. Nie, et al., Chem. Commun. 51(2015) 16417-16420; (d) L. H. Wang, Z. J. Zhang, H. Y. Zhang, H. L. Wu, Y. Liu, Chin. Chem. Lett. 24(2013) 949-952; (e) A. Singh, W. T. Yip, R. L. Halterman, Org. Lett. 14(2012) 4046-4049; (f) O. Buyukcakir, F. T. Yasar, O. A. Bozdemir, B. Icli, E. U. Akkaya, Org. Lett. 15(2013) 1012-1015. |
| [10] |
K. Kim, Chem. Soc. Rev. 31(2002) 96-107. DOI:10.1039/a900939f |
| [11] |
V. Kolman, M.S. Khan, M. Babinský, R. Marek, V. Sindelar, Org. Lett. 13(2011) 6148-6151. DOI:10.1021/ol2023888 |
| [12] |
H. Yang, Y.L. Liu, K. Liu, et al., Langmuir 29(2013) 12909-12914. DOI:10.1021/la4025102 |
| [13] |
V. Sindelar, S. Silvi, A.E. Kaifer, Chem. Commun. 20(2006) 2185-2187. |
| [14] |
(a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam, B. Z. Tang, Rev. 115(2015) 11718-11940; (b) J. S. Yang, C. K. Lin, A. M. Lahoti, et al., J. Phys Chem. A 113(2009) 4868-4877. |